癌転移のメカニズム。癌関連遺伝子の二つの癌関連遺伝子を最初に癌遺伝子に突然変異させるのはヘルペスウイルスです。更に癌細胞を転移させるきっかけとなる癌原発巣である癌細胞塊の塊から一個の癌細胞が塊からはがれるのはすべてのヘルペス感染細胞が隣接する細胞同士の接着性の喪失(loss of adherence)から始まるがん浸潤が起こり始めるのががん転移の始まりなのです。
転移癌を作るのも癌細胞を最初に癌化させて癌を発がんさせて癌細胞の中で増殖した膨大な数のヘルペスが次々と隣の細胞に細胞膜を破って感染するには頑丈な細胞と細胞の接着を切り離す必要があります。新たに感染した数多くの細胞も癌遺伝子を徐々に徐々に癌化させられていくのにも時間がかかります。ところが100万個の癌細胞集塊の中の一個の癌細胞が癌細胞集塊から離れて行く浸潤から癌の塊からやっと100%剥離された一個の癌細胞が転移を始めます。次々と連続的に癌細胞がヘルペスの感染がもたらす細胞変性効果が影響を発揮させ癌細胞塊からの癌細胞の剥離が増えていくのです。つまり原発巣の一個の癌細胞がヘルペスのよって生まれ増殖して隣の細胞を巻き込んで癌化させて集団になって初めて100万個の癌細胞塊から一個の癌細胞が転移するメカニズムを解明しましたので説明しましょう。細胞変性効果(Cytopathic effect略して CPE)とは何でしょうか?細胞変性効果は、ヘルペスウイルスの侵入によって引き起こされる宿主細胞の形態変化のことです。ヘルペスウイルス感染により、宿主細胞が溶解する場合や、細胞の複製が阻害され溶解せずに細胞が死滅する場合もある。これらの現象はどちらもCPEが原因で発生します。ヘルペスウイルスが宿主細胞にこれらの形態学的変化を引き起こす場合、ヘルペスウイルスが細胞変性性を持っているといいます 。CPEがヘルペスウイルス感染によって生じる細胞の形態的な変化には、球状化(rounding)、収縮(shrinkage)、屈折性の増加(increased refractility)、細胞の融合(fusion)、凝集(aggregation)、接着性の喪失(loss of adherence)、溶解(lysis)が挙げられています。これらの現象が様々に組み合わさり、殺し切れないヘルペスウイルスに特徴的な変化が永続的にもたらされるのです。
このherpesの他の細胞への感染と癌細胞の転移との関係の解明も世界で初めての発見です。何故わたしの松本医学には世界で最初の発見が多いのでしょうか?それは現代に最後に残されたすべての病気の原因はワクチンが永遠に作れない従って殺しきれない病原体はヘルペスしか残されていないのでヘルペスは免疫が落ちた時に堂々とあらゆる組織の細胞で増殖し免疫が回復すると再び増えたherpesとの戦いがあらゆる組織の細胞内や細胞外で行われることを知っているのは私だけだからです。いや!本当は世界中の偉い医学者で知らない人はいませんが!!!!!
癌の転移とは何でどのようにして転移や浸潤や播種が起こるのでしょうか? 癌の転移とはがん細胞が最初に発生した場所(原発巣)から、播種したり血管やリンパに入り込み、血液やリンパの流れに乗って別の臓器や器官に移動し、そこで増えることです。癌の播種とは癌のできた臓器からがん細胞がはがれ落ち、近接する体内の空間(胸腔や腹腔)に散らばるように広がることをいいます。いずれにしろの癌細胞塊集団から癌細胞が剥がれて原発巣から離脱し始めるのは転移のし始めなのです。癌細胞が原発巣に限局する時の治癒率は改善してきているのですが遠隔転移が形成された転移がんの予後は依然として極めて不良なのです。何故原発巣に限局した癌と転移癌治癒の違いがそんなに大きいのでしょうか?ここで注意しておきたいのは癌治癒か癌の完治ではないことです。癌の根本治療ではないことです。つまり癌の根本の原因はヘルペスですから癌の完治は癌細胞にはherpesが細胞質にも核の染色体にも隠れ住んでいますからロイアルレイモンド博士の「光癌療法」で癌ウイルスであるヘルペスウイルスを殺せば癌細胞もろとも癌原ウイルスであるherpesウイルスも殺しきることができ、ついでにこの世にない自己免疫疾患も治せるし起こらないようにもできます。ロイアルレイモンド博士の新しく書き直されたTHINKER logoの「光癌療法」の記事はこの論文のすぐ後に掲載していますからいつでも読めます。従ってロイアルレイモンド博士の「光癌療法」の装置は簡単に作れるので「転移がどのように起こるか」詳しく説明するのは面倒なことですがヘルペスが他の細胞に感染する転移(?)を起こすきっかけを作るのは、細胞の中でherpesが大量に増殖したherpesが細胞の膜を破り癌細胞同士の細胞と細胞の強い細胞同士の接着結合を切り離して癌細胞塊から播種し始めるメカニズムともう一つ癌細胞と細胞マトリックスとの結合を切り離して血流や組織やリンパ流に入り込んで遠隔転移をやり遂げるかを詳しく説明していきます。
癌転移の種類はいくつありますか?癌の転移は癌を作ったヘルペスウイルスが癌細胞を手引きして、大きく5種類の転移を起こすのです。血管を通って転移する①「血行性転移」、リンパ管を通って転移する②「リンパ行性転移」、種を蒔くようにがん細胞が広がっていく③「播種性転移」、隣接する臓器に広がっていく④「浸潤」です。⑤管腔性転移の五つです。遠隔転移とは何でしょうか?がん細胞が最初に発生した場所(原発巣)から、血管やリンパ管に入り込み、血液やリンパの流れに乗って別の臓器や器官に移動し、そこで増殖することをいいます。 肺や肝臓、脳、骨など血液の流れが豊富な場所や、リンパの流れが集まる場所であるリンパ節に遠隔転移することが多いのです。播種性転移とは何でしょうか?種を蒔くようにがん細胞が散らばっていくことからつけられた名前です。 内臓と腹膜の間に腹腔があり、胸膜同士の間に胸膜という隙間があります。 この隙間に、近くに出来た臓器にあるがんが増殖して、その内面に種を蒔くように広がっていくのが播種性転移です。癌浸潤とは何ですか?がんが原発巣、つまり最初に発生した部位から周囲の組織や臓器へと広がっていく現象を指します。 がん細胞は初めは小さな塊として存在しますが、時間が経つにつれて徐々に大きくなり、隣接する癌組織が大きくなりその結果、広がることになります。管腔性転移とは何でしょうか?経管腔性転移とか経気道性転移とも言います。主に肺にできたがんが、気道の中を空気の流れに乗って肺のほかの部分にたどり着く経路を通じての転移です。以上述べたように五つの固形癌の転移が起こるのは単純に転移が起こるのではなくその背後には知られていない固形癌の転移の分子機構があるのでその分子機構を明らかにしたいのでこの論文を作成し始めたのです。
癌の転移研究の歴史から始めます。1986年に転移のひとつである「浸潤」がThree Step Theoryという3段階説が提唱されました。癌の浸潤は一段階目は癌細胞と基底膜の接着の乖離、二段階目は基底膜成分の分解、三段階目は癌細胞の運動の三つが絡んでいる3段階説が出て転移のメカニズムを癌細胞と細胞同士の接着を分子レベルで研究され始めました。その研究の結果、①癌細胞はすべて同質の癌という病気と考えられた時代もあったのですがその後、癌は生物学的に同質ではなく不均一であるとわかりました。これは癌細胞の種類によって細胞一つに23500種類の遺伝子の中にある800種類のがん関連遺伝子のどの遺伝子がヘルペスによって癌化したかによって癌細胞が作る蛋白が不均一であるからです。②癌細胞は絶えざるヘルペスによる遺伝子突然変異によってヘルペスが増殖しやすいかつ転移しやすい細胞形質に代わりごく少数の転移しやすい、つまり癌細胞が細胞同士を結合させる接着能がヘルペスが低下させて癌細胞が増えて転移しやすくなっていくのです。③転移のすべての過程で様々な接着因子、蛋白分解酵素、癌増殖因子、血管新生因子、ケモカインなどの多くの分子が関与しているのです。がんの転移は以上の複雑なヘルペスによって誘導された転移の過程を乗り越えた癌細胞のみ数百万個に一個の癌細胞のみ転移が可能となるのです。
以上述べたように5種類の転移様式がありますがその転移のメカニズムは基本的にはあらゆる細胞に感染したヘルペスが細胞の核の染色体のゲノムの遺伝子に細胞が分裂する時に23本の染色体も分裂して46本の一倍体の染色体になるときにヘルペスのゲノムがその46本の一倍体の染色体を切り離して46本のゲノムに自由自在に組み込まれ細胞の遺伝子の組み換えを起こしてしまうと遺伝子の突然変異が必ず起こります。たまたまその突然変異が二種類の癌関連遺伝子である癌原遺伝子とがん抑制遺伝子であれば一個の癌細胞が生まれるのです。勿論この時ヘルペスによって細胞の正常な遺伝子のDNAが組み換えという異常な損傷を受けると遺伝物質DNAの主な活動のひとつである修復が始まります。ヘルペスが組み替えてしまった遺伝子をもとの戻すための修復は元の誘導したものですから遺伝子に組み換えればいいのです。この組み換えの遺伝子は修復の遺伝子と同じです。ヘルペスによってつくられたDNA損傷は塩基損傷とDNA鎖切断の二つに区分されます。細胞に備わったヘルペスによる損傷の修復系はあらゆる種類のDNA損傷を事実上100%治癒する力があるのでヘルペスによる二種類の癌関連遺伝子である癌原遺伝子とがん抑制遺伝子が損傷されてもそう簡単には癌細胞は生まれないのです。しかもヘルペスウイルスが自分のゲノムを48本の一倍体のどの遺伝子の組み換えをやるかどうかはすべて偶然なのです。従って癌になる一番大きなファクターはストレスのために免疫を抑えすぎてherpesをどれだけ増やしたかですが同じ数のヘルペスに感染されていても癌になるかは偶然であり運の問題だけなのです。因みにherpesが感染細胞の遺伝子のDNAを組み替えるやり方を部位特異的組み換えと言います。
5種類の転移のメカニズムは基本的には似ていますから血行性転移とリンパ行性転移の二つの脈管性転移に力点を置いて説明します。転移は六段階の過程から成り立っています。①癌細胞の原発巣での増殖②原発巣から癌細胞の離脱と脈管への浸潤③脈管内での移動④転移臓器の血管内皮細胞への接着⑤転移臓器への浸潤⑥転移臓器内での増殖の6つの過程が連続した結果、生じるのです。又すべての6つの過程において癌細胞は人体の免疫排除機構から逃れなければなりませんが果たして人体の細胞は異物ではありませんから免疫には敵と認識されません。癌細胞は突然変異が二種類の癌関連遺伝子である癌原遺伝子とがん抑制遺伝子で生じても癌細胞は二種類の癌関連遺伝子が作る蛋白だけが正常な細胞とは異なるだけですから異常な蛋白質ができてもそのために癌細胞自体が異物と免疫に認識されるかどうかの問題が残ります。私の答えはNOです。その根拠は遺伝子疾患は免疫によって排除されないという厳粛な事実があるからです。それは遺伝子は作るべき蛋白を指定しています。ところが遺伝子病の患者さんは正常でない蛋白質を遺伝子病の異常な蛋白質を作ったからその蛋白質のために免疫はその細胞を異物として排除してくれますか?NOです。従って癌も遺伝子病も免疫の排除は一切関りがないのです。しかし癌を作るのはヘルペスウイルスですからそのherpesを排除する働きは又別問題です。
現代の病気の原因は免疫では絶対に殺し切れない従って一度感染してしまってもワクチンの効果に必要な免疫記憶細胞もできないのでワクチンも人工的には効果がなくしかも免疫が高いときは宿主細胞の核の染色体のゲノムの潜伏感染するときに染色体の遺伝子であるDNAを切断してヘルペス自身のゲノムをランダムに組み込みかつ遺伝子の組み換えを自由自在にやってしまい遺伝子を突然変異を起こし癌も起こし原因不明な遺伝子病も作りあらゆる疾患を起こしてしまうのです。しかも患者の免疫が落ちると細胞が二つに分裂するときに結合していた2倍体の23本の染色体が父親と母親の一倍体の染色体の46本になったときにどの染色体に自由に遺伝子を組み込みDNAを部分特異的組み換えによって遺伝子を突然変異させてしまうのです。このように細胞が分裂する時にのみ利用したり新たに作る酵素やタンパク質や機能をヘルペス自身も利用したり奪い取ったりしてヘルペスが持っていない細胞の増殖機構を活用しながらヘルペス自身も何百倍、何千倍にも増えていくのです。このために細胞自身は自分の分裂や2倍の大きさになるために必要な蛋白質や細胞小器官も作れなくなったりするとヘルペスの溶解感染といわれる再活性化とも言われる現象が起こり正常な遺伝子の発現が起こらなくなり細胞変性効果といわれる細胞の変性による形態の変化が生じてしまい様々な細胞機能障害を起こし細胞尾複製ができなくなり最後は細胞壊死になって死んでしまうのです。細胞変性効果(Cytopathic effect略して CPE)とは何でしょうか?細胞変性効果は、ウイルスの侵入によって引き起こされる宿主細胞の形態変化のことです。ヘルペスウイルス感染により、宿主細胞が溶解する場合や、細胞の複製が阻害され溶解せずに細胞が死滅する場合もあります。溶解感染とはヘルペスウイルスを産生している感染のことで子孫ウイルス産生およびウイルス伝播も起きます。EBウイルス溶解感染では約 80 個のウイルス遺伝子が秩序立って発現し,短時間の内にウイルスゲノム複製と,続く粒子形成が協調的になされます。細胞が解けて死んでしまうことではありません。溶解感染も壊死感染もCPE(細胞変性効果)が原因で発生します。
ヘルペスウイルスが宿主細胞にこれらの形態学的変化を引き起こす場合、細胞変性性があると言われ、ウイルスの感染により細胞に生じる変化には、球状化(rounding)、収縮(shrinkage)、屈折性の増加(increased refractility)、融合(fusion)、凝集(aggregation)、接着性の喪失(loss of adherence)、溶解(lysis)が挙げられています)。これらの現象が様々に組み合わさり、各ウイルスに特徴的な変化がもたらされると考えられています。
Epstein-Barrウイルス(Epstein-Barrvirus;EBV)は,γ-ヘルペスウイルス亜科に属するDNAウイルスで,最初に発見されたヒト癌ウイルスでもある.EBVは潜伏感染と溶解感染(ウイルス産生感染)の2つの感染様式を持ち,基本的にEBV感染細胞は潜伏感染を呈するが,ときに溶解感染へと移行し,子孫ウイルス産生およびウイルス伝播が起きる.溶解感染では約80個のウイルス遺伝子が秩序立って発現し,短時間の内にウイルスゲノム複製と,続く粒子形成が協調的になされる.本稿では,潜伏感染から溶解感染へと変化するときに,宿主細胞内でどのような変化が起きているのかについて,筆者の研究経緯を交えながら概説する.
ウイルスは宿主細胞内で短時間に爆発的に増殖し,感染細胞あたり数百から数万の子孫ウイルスが産生される.この非常に効率的な増殖には,時間と場所の制御が必須である.例えば,必要な“時”に必要な“もの(遺伝子産物)”が必要な“量”だけ提供可能となる秩序だったウイルス遺伝子発現や,反応に必要な因子が密に存在し,反応速度を最大化する“場”である.さらに,ウイルスは宿主細胞にとって異物であるため,宿主細胞は侵入した異物(ウイルス)を排除しようとする.これを巧みに回避する機構もウイルスは有している.このように,ウイルスは多重かつ多階層的に宿主細胞内の環境をコントロールしながら,宿主細胞内で増殖する.この感染細胞内で生じるダイナミックな変化を理解することは,抗ウイルス戦略を考える上で非常に重要である.
EBVについてEpstein-Barrウイルス(EBV)は,γ-ヘルペスウイルス亜科に属するDNAウイルスで,成人の約9割が抗体陽性とされ,最も広く浸淫しているウイルスのひとつである.EBVの初感染は多くは無症候性であるが,思春期以降では伝染性単核症の原因となる.一方で,EBVはヒト腫瘍ウイルスでもあり,バーキットリンパ腫や上咽頭癌,胃癌などEBV関連腫瘍の新規発症者は全世界で年間200,000人にものぼる.
EBVはTリンパ球や上皮細胞にも感染するが,自然宿主はBリンパ球である.EBVが感染したBリンパ球はリンパ芽球へと形質転換し,分裂増殖を繰り返すようになる(不死化する).このとき,感染細胞ではごく限られたウイルス遺伝子産物しか発現しておらず,ウイルス粒子産生もなく,EBVは潜伏感染状態となる.そのため,EBVはヘルペスウイルスの潜伏感染モデルとして研究されてきた.
潜伏感染と溶解感染EBVは潜伏感染と溶解感染の2つの感染様式を持ち,基本的にEBV感染細胞は潜伏感染を呈する.そして,ときに溶解感染へと移行し,子孫ウイルス産生およびウイルス伝播が起きる.潜伏感染細胞は分裂増殖するため,感染の維持には娘細胞にウイルスゲノムが分配される必要がある.潜伏感染では,ウイルスゲノムは宿主の染色体複製装置によりS期に1回複製される.複製されたウイルスゲノムは,EBNA1を介して宿主染色体と結合し細胞分裂時に宿主染色体と共に分配される.EBNA1を介した分配機構は,娘細胞に等しくウイルスゲノムを分配することを可能にし,ウイルスゲノムは感染細胞内で維持され続ける.潜伏感染細胞ではウイルスゲノムは環状DNAとして維持される.一方,溶解感染では短時間で大量のウイルスゲノムを複製する必要があるため,ウイルスゲノム複製は7種のウイルス複製蛋白質から構成されるウイルス複製装置により実行される.ローリングサークル型の複製が起き,中間産物として長いhead to tail concatemerができる.これは後に,ユニットサイズに切断され,カプシド内へパッケージされる.
溶解感染への移行に伴う細胞内環境変化潜伏感染から溶解感染への移行は,BZLF1タンパク質の発現により規定される.BZLF1タンパク質は,ウイルスゲノムの複製開始部位に結合してウイルスのコードする複製タンパク質群の会合を誘導するとともに,転写因子としても機能して溶解感染関連ウイルス遺伝子群(詳細は後述する「ウイルス遺伝子転写カスケード」の項を参照)の発現を誘導する.さらに,BZLF1タンパク質はp53などの宿主タンパク質の溶解感染の進行に応じた緻密な制御にも関わる.
溶解感染ではウイルス粒子産生に細胞内資源を集中させるため,EBVは細胞周期もコントロールする.溶解感染では,ウイルスゲノム複製に適したlate-G1からS期の環境に細胞内環境が整えられる.BZLF1タンパク質は低リン酸化状態(活性化状態ではない)のp53とDNAとの結合を増強することで,p53の下流にある細胞周期調節因子p21Cip1/Waf1の発現を誘導し,溶解感染の初期には細胞周期をG1期付近に止める.ウイルスゲノム複製が爆発的に行われると,ウイルスゲノムは異常DNAとして検知され,宿主DNA損傷応答が誘導される.なお,EBVに限らず,ウイルスゲノム複製で宿主DNA損傷応答が惹起されることは様々なウイルスで報告されている.宿主DNA損傷応答はゲノムの完全性(genome integrity)を維持するための機構で,傷害DNAは取り除き,修復される.修復が困難な場合には細胞死が誘導される.この宿主DNA損傷応答でもp53は中心的な役割を果たすが,ウイルスにとってはp53の活性化による細胞死の誘導は不都合である.そのため,これを巧みに回避する機構をEBVは有している.溶解感染で宿主DNA損傷応答が惹起されると,ATMがリン酸化し,下流のChk2へとシグナルを伝える.これらのキナーゼはp53をリン酸化し,活性化状態にするが,溶解感染の中期以降ではp53の下流にシグナルが伝達されない.これは,p53がE3ユビキチンリガーゼの一種であるElongin B/C-Cul2/5-SOCS-box protein(ECS)複合体とBZLF1タンパク質を介して結合し,p53がユビキチン化され,分解されるためである.興味深いことに,BZLF1タンパク質を介したECSリガーゼ複合体との結合は,Chk2によるp53のC末端のリン酸化により増強する.従って,EBVは活性化状態のp53(ウイルスにとってアポトーシスへと導く可能性がある危険なp53)を優先的に分解することが可能となる.
一方で,宿主DNA損傷応答はウイルスにとって不都合な宿主反応とは限らない.それは,ウイルスゲノムの完全性の維持および効率的なウイルスゲノム複製に,EBVは宿主DNA損傷応答をも利用しているからである.ATMからのシグナルはMRN複合体へと伝えられて,相同組換え修復酵素をウイルスゲノム上にリクルートする.これらの複合体により,ウイルスのDNAポリメラーゼによって複製された不完全なゲノムが補完され,効率的なウイルスゲノム複製が達成される.このように,宿主DNA損傷応答はウイルスにとって重要である.これをさらに確実なものにするため,EBVは宿主DNA損傷応答を増幅する機構も備えている.その中心を担うのが,EBVのコードする唯一のプロテインキナーゼBGLF4タンパク質である.BGLF4キナーゼは,ヒストンH2AXをリン酸化し,宿主DNA損傷応答を増幅させる.
BZLF1タンパク質と同様に,BGLF4キナーゼも溶解感染のための細胞内環境調整に多面的に機能する.BGLF4キナーゼはコンデンシンやトポイソメラーゼIIをリン酸化し,宿主染色体を凝集させる30).さらに,宿主の染色体複製開始に関わるMCM複合体のMCM4のリン酸化を介して,MCM複合体のヘリカーゼ活性を抑制する.そのため,溶解感染では宿主のゲノム複製が抑制され,ウイルスゲノム複製のみが実施される.BGLF4キナーゼは細胞周期調節因子p27Kip1のThr-187をリン酸化する.リン酸化されたp27Kip1はSCFSkp2によって,ポリユビキチン鎖を付加され,分解へと誘導される.これも,溶解感染でのS期CDKの活性化に貢献する.さらに,BGLF4キナーゼは,細胞内のdNTPの量を制御する因子であるSterilealphamotifandHDdomain1(SAMHD1)をリン酸化することが最近,報告された.リン酸化によりSAMHD1のdNTPase活性は低下し,新たに複製されるゲノムの原料となるdNTPプールが増加することで,効率的なウイルスゲノム複製が可能となる.加えて,BGLF4キナーゼは核膜の裏打ちタンパク質であるラミンA/Cのリン酸化を介して,核膜構造の再構成を促し,EBVカプシドのnuclear egressにも関与する.また,BGLF4キナーゼは詳細なメカニズムは未だ不明であるが,後期遺伝子転写を正に制御することが報告されている.
多機能ウイルスタンパク質による宿主タンパク質の制御が幾重にも重なってウイルス複製に適した環境が形成されていく.このように整えられた細胞内環境は,S期CDKs(Cyclin A-およびCyclin E-CDKs)は高い活性を示すが,宿主のゲノム複製は起きないというS期に類似した状態でS期様環境と呼んでいる.
ウイルス遺伝子転写カスケードEBVの溶解感染では,ほぼすべてのウイルス遺伝子が秩序立ったカスケードによって発現する.まず,転写因子をコードしている前初期遺伝子BZLF1とBRLF1が発現し,溶解感染が開始する.BZLF1タンパク質とBRLF1タンパク質は,ウイルスゲノム複製に必要なウイルス遺伝子群(初期遺伝子)の発現を誘導する.初期遺伝子産物がウイルスゲノムを複製すると,新規に合成されたウイルスゲノムを鋳型として,ウイルス粒子形成に必要な遺伝子がコードされている後期遺伝子が発現する.
S期様環境はウイルス遺伝子発現にも適した環境である.S期CDK活性が高い状態であるため,Rbは高リン酸化状態となり,E2F転写因子が遊離する.E2F-1は宿主の複製タンパク質の発現を誘導するとともに,EBVのDNAポリメラーゼを含む初期遺伝子の転写も誘導する.E2F-1は宿主DNA損傷応答によっても活性化するため,EBV溶解感染では少なくとも2つの制御系により活性化されている.
さらに,S期CDK活性は後期遺伝子発現の時間的な制御にも関わることを最近報告した.EBVを含むβ-およびγ-ヘルペスウイルスではウイルス後期遺伝子の転写が,ウイルスのコードする転写調節因子複合体(viral preinitiation complex;vPIC)によって制御される.このvPIC複合体の構成因子であるBDLF4は,S期CDKsの基質であり,ウイルス複製の進行とともにBDLF4は高度にリン酸化される.低リン酸化状態のBDLF4は不安定で,すぐにユビキチン化され,分解されてしまう.しかし,BDLF4はリン酸化により安定化し,vPICが形成可能となる.そのため,溶解感染の後期にBDLF4が安定化し,後期遺伝子の発現を誘導するという時間的な制御が可能となる.このように,EBVは溶解感染の進行に伴う宿主環境の変化を巧みに利用し,ウイルス遺伝子発現の時間的な制御を行っている.
ウイルス遺伝子発現は時間的な制御だけでなく,空間的な制御も受けている.溶解感染細胞の核内にはReplication compartmentと呼ばれる核内構造体が出現し,ウイルス複製工場として機能する.Replication compartmentには複製と転写に関わる宿主タンパク質とウイルスタンパク質が高密度に局在する.溶解感染の初期には核内に点在するように存在するReplication compartmentは,ウイルス産生に伴って,最終的には核内の大部分を占めるまで成長する.免疫染色と3次元再構成解析により,Replication compartmentはウイルスタンパク質BMRF1で形成されるコア構造が存在し,コア構造の内と外でウイルス前期遺伝子と後期遺伝子の転写がそれぞれ起こることを明らかにした.Replication compartmentは膜構造を持たないオルガネラ様の構造体であるが,その形成には液–液層分離が関与していることも明らかになりつつある(筆者ら未発表データ).したがって,EBVはウイルス遺伝子の転写を時空間的に制御することで,効率的な子孫ウイルス産生を達成する.
須であるが,溶解感染関連遺伝子(溶解感染時に発現するウイルス遺伝子)の一部は,ウイルス産生時の他にも発現し,機能を果たしている.例えば,EBVがBリンパ球へ初感染する場合にも一部の溶解感染関連遺伝子が発現し,感染細胞のアポトーシスを抑制していることや,潜伏感染の成立までの間に不完全な溶解感染を経由すること(筆者ら未発表データ)が明らかとなっている.また,組換えEBVを使用したマウス実験で,個体でのウイルス発がんは潜伏感染だけでなく,溶解感染も関与することが示され,臨床検体を使用したEBV関連腫瘍の次世代シーケンス解析でも溶解感染(特に不完全な溶解感染:ウイルス複製は起きるが粒子形成が完結しない)の重要性が示唆された.したがって,溶解感染による細胞内の環境変化はウイルス産生に留まらず,EBV関連疾患と密接に関連しているかもしれない.さらに,組織中でのウイルス感染を考えると,感染細胞の隣には必ず非感染細胞(未感染細胞)が存在し,それらの相互作用もあるはずである.実際に,EBVのコードする癌原性タンパク質LMP1は発現細胞と非発現細胞との間で細胞競合(適応度の高い細胞集団が適応度の低い細胞集団を排除する現象)が観察された.したがって,今後は細胞内での環境変化に加えて,細胞外の変化についても解析し,疾患発症や感染の成立を含めた感染細胞の運命決定機構について研究していきたい.さらに,個体レベルでウイルス感染を考えると,EBVが単独で感染していることは稀で,他のウイルスと宿主個体の中で共存することになる.そこに何らかの相互作用が生じることは想像に難くない.そのため,異種ウイルス間の相互作用にも迫る研究を展開していきたいと考えている.
それではどのようにしてヘルペスウイルスは宿主の癌関連遺伝子を突然変異させて癌化させて癌細胞を生み出すのでしょうか?ヘルペスは感染細胞の核の染色体のDNAに潜伏感染するときには一対のDNAの鎖を切ってヘルペスウイルスのゲノムを組み込んで細胞のヌクレオチドの配列を組み替えてしまうのです。このようなヘルペスウイルスによる宿主のDNAに入り込むときにつかうDNA組み換えを部位特異的組み換えといいます。この遺伝子の部位特異的組み換えによってたまたま2種類の癌関連遺伝子の配列が変わってしまうと一個の癌細胞が生まれてしまうのです。この時細胞のDNA修復機構が部位特異的組み換えを正常なもとの遺伝子配列に修復できなければ確実に一個の癌細胞が生まれてしまうのです。
ヘルペスウイルス
(HEF細胞) 線維芽細胞 (HEF)
ウイルスが増殖し、すでにすべての細胞が破壊された状態。
herpesvirus
ヘルペスウイルス
(VERO細胞) ベロ細胞 はアフリカミドリザルの腎臓上皮細胞に由来する、細胞培養に使われる細胞株である。HeLa細胞と並んで最もよく使われている細胞株の一つである。
丸みをおびた細胞変性像があちこちに散在している。間はまだウイルスが増殖していない正常細胞。
CMV
サイトメガロウイルス
(HEF細胞)
膨らんだ、透明感の高い細胞変性像が細胞から細胞へのゆっくりと拡大していくのが特徴。
ヘルペスが起こす最も大きな問題は宿主の細胞の核のゲノムに自分のゲノムを組み込んだ後細胞のDNAを部分特異的組み換えを起こすときに癌関連遺伝子の2種類を突然変異させてがん細胞に仕立て上げてしまうことです。
細胞変性効果
細胞変性効果(さいぼうへんせいこうか、Cytopathic effect: CPE )は、ウイルスの侵入によって引き起こされる宿主細胞の形態変化のこと。ウイルス感染により、宿主細胞が溶解する場合や、細胞の複製が阻害され溶解せずに細胞が死滅する場合がある。これらの現象はどちらもCPEが原因で発生する。ウイルスが宿主細胞にこれらの形態学的変化を引き起こす場合、細胞変性性がある(cytopathogenic)と言われる。CPEは光学顕微鏡下で容易に観察でき、その変化として感染細胞の円形化、収縮、集合、膨化、崩壊、隣接細胞との融合による合胞体(多核巨細胞)形成、核または細胞質封入体の形成などがある。
CPEやその他の細胞形態の変化は、細胞致死性ウイルスによる多くの影響のうちのごく一部である。細胞致死性ウイルスが感受性細胞に感染すると、ウイルスは、細胞形態や、細胞の生理機能、続く生合成機構の変化を通じて宿主細胞を死滅させる。これらの変化は、効率的なウイルス複製に必要であるが、宿主細胞が犠牲になる。
CPEはウイルスおよび感染した細胞の種類によって特徴的であることが多いので、ウイルス種の推定に利用できる。また、TCID50により、ウイルスの量(正確にはウイルス感染価)が測定できる。
細胞変性効果(円形化)培養フラスコの底に敷石状に生育している培養細胞がウイルスの感染によって円く変形し、やがてフラスコからはがれてプラーク(空隙、写真中央)を形成する。
細胞変性効果(合胞体)敷石状に生育した培養細胞同士がウイルス感染によって細胞膜の融合を起こし、細胞核が中央に凝集して(写真中央)多核巨細胞様の形態になる。
診断
CPEは、ウイルス感染の診断において重要な判断要素である。多くのCPEは、光学顕微鏡の低倍率で、コンデンサーを下げ、虹彩絞りを部分的に閉じた状態で、固定されていない、染色されていない細胞で観察できる。ただし、CPEの一種である封入体では、細胞を固定して染色し、光学顕微鏡で観察する必要がある。一部のウイルスのCPEは特徴的であるため、感染した動物や人間を診断する際の重要なツールになることがある。CPEの出現率も、ウイルスの種類を特定するための重要な特性である。感染多重度が低い状態でin vitroで4〜5日後にCPEが出現した場合、ウイルスの増殖は遅いと見なされる。 感染多重度が低い状態でCPEがin vitroで1〜2日後に出現する場合、ウイルスの増殖は速いと考えられる。感染多重度が高い場合、CPEは急速に発生するため、診断においては接種は常に感染多重度を低くする。
通常、ウイルス感染の最初の兆候は細胞の円形化である。その後、封入体が宿主細胞の細胞核や細胞質に現れることが多い。封入体は、最初に、患者の血液塗抹標本または感染組織の染色切片の光学顕微鏡検査によって識別できる。ただし、封入体の組成を完全に調べるには、電子顕微鏡検査を行う必要がある。封入体は、ウイルス複製副産物の蓄積か、宿主細胞の細胞小器官や細胞構造の変化のいずれかの可能性がある。
一部のウイルス感染は、奇妙なCPEである合胞体の形成を引き起こす。合胞体は、多くの核を含む大きな細胞質の塊であり、通常、感染した細胞の融合によって生成される。この機構によりウイルスが感染細胞から非感染細胞に広がることができるため、ウイルスにとって有利に働く。
ウイルス感染は、臨床的に関連する表現型のCPEを持っている可能性がある。たとえば、 C型肝炎ウイルス(HCV)の場合、脂肪肝はウイルスの遺伝子型や遺伝子組成の特定に利用できる。 HCV遺伝子型3の患者は、遺伝子型1の患者よりも肝脂肪症を発症する可能性が有意に高いと言われている。また、CPEは、新薬の有効性を判断するための研究にも利用され、例えば細胞の生存率を評価するために、デング熱ウイルスのCPEをスクリーニングするアッセイが開発されている。
CPEの宿主細胞特異性により、CPEを使用して実験上の宿主の違いを確認することができる。多くのウイルス感染では、さまざまな宿主細胞株が特徴的な反応を示すことがある。現在、細胞株の信頼性と純度について、研究コミュニティ内で多くの懸念があり、研究所内および研究所間で汚染が増加しているが、特定の細胞株の純度を確認するためにCPEが利用できる。たとえば、 HeLa CCL-2は、さまざまな研究分野で使用される一般的な細胞株であるが、HeLa細胞の純度をテストするために、コクサッキーウイルスB3の接種後に発生する形態変化や細胞の死亡率などのCPEが利用される。Carsonらは、この違いは、ラボで何世代にもわたって増殖してきたHeLa細胞の均質性と比較して、市販のHeLa細胞の不均質性に起因すると判断した。
CPEの種類
完全剥離
宿主細胞単層の完全剥離は、最も深刻なタイプのCPEである。観察には、細胞をガラス表面に播種し、宿主細胞のコンフルエントな単層を形成したあと、ウイルス感染を行う。単層のすべての細胞は急速に収縮し、核濃縮と呼ばれるプロセスで高密度になり、3日以内にガラスから剥がれる。この形式のCPEは、通常、エンテロウイルスで見られる。
部分剥離
宿主細胞単層の部分剥離は、全剥離ほど深刻ではないが、全剥離と同様に、宿主細胞をガラス表面に播種してコンフルエントな単層を形成したあと、ウイルス感染を行うことで観察される。部分剥離は、単層内の一部の細胞の剥離が特徴であり、一部のトガウイルス、一部のピコルナウイルス、および一部のパラミクソウイルスで一般的に観察される。
局所変性
局所変性は、宿主細胞単層の局所的な攻撃により生じる。このタイプのCPEも最終的には組織全体に影響を与える可能性があるが、初期段階と拡散段階はフォーカス(foci)と呼ばれる局所的なウイルスの感染中心で発生する。局所変性は、細胞外の培地を介した拡散ではなく、ウイルスの直接的な細胞間移動が原因である。この移動の様態により、完全剥離や部分剥離と異なる特徴的な局所効果を引き起こす。最初に、宿主細胞は肥大し、円形化し、屈折する。最終的に、宿主細胞は表面から剥離する。ウイルスの拡散は同心円状に発生するため、持ち上がった細胞の周りには肥大し円形化した細胞があり、その周りを健康な組織に取り囲んでいる。このタイプのCPEは、ヘルペスウイルスとポックスウイルスに特徴的である。
膨潤と凝集
膨潤と凝集は、宿主細胞が著しく膨潤するCPEである。細胞が拡大するとクラスター状に凝集するが、最終的には細胞は非常に大きくなり、分離する。このタイプのCPEは、アデノウイルスに特徴的である。
泡状変性
泡状変性は空胞化とも呼ばれ、大きく多数の細胞質液胞の形成による。このタイプのCPEは、宿主細胞の固定と染色でしか観察できない。泡沫変性は、特定のレトロウイルス、パラミクソウイルス、フラビウイルスに特徴的である。
合胞体
合胞体は、細胞融合およびポリカリオン形成としても呼ばれ、4つ以上の宿主細胞の原形質膜が融合し、少なくとも4つの核を持つ拡大した細胞が生成されたものである。大きな細胞融合は染色せずに見えることもあるが、このタイプのCPEは通常、宿主細胞の固定と染色により検出される。ヘルペスウイルスは、細胞融合や他の形態のCPEを特徴的に形成する。一部のパラミクソウイルスは、このCPEのみを形成するため、細胞融合の形成により同定されることがある。
封入体
封入体(核内または細胞質内の不溶性の異常構造)は、宿主細胞の染色像が変化した領域を指すことから、染色でのみ見られる。通常、宿主細胞内でウイルスタンパク質や核酸が合成されている領域や、ビリオンが組み立てられている領域である。場合によっては、封入体は活性ウイルスがなくとも存在し、ウイルスの瘢痕であることもある。封入体はウイルス株によって異なり、数や大小はさまざまで、形状も円形や不規則であったりする。また、核内、細胞質内のいずれにも形成され、染色性も好酸球性、好塩基性とさまざまである。
一個の癌細胞が10年かけて1センチの大きさの癌細胞塊に10億の癌細胞にまで増殖するという理論は100%間違っています。何故でしょうか?癌細胞は1年間に3回分裂するので10年たてば分裂するたびに二倍、二倍と30回分裂するので230=1073741824となり約10億7374万なのです。
本当に癌化した癌染色体だけが勝手に増えるのか?
がんの転移の最初の浸潤(Invasion)について詳しく説明します。
英語から翻訳-浸潤は、がん細胞が直接伸びてがん内の隣接組織に侵入するプロセスです。これは一般に、循環系またはリンパ系を通ってより離れた場所へがん細胞が広がる転移とは区別されます。しかし、リンパ管浸潤は一般に転移の最初のステップです。 細胞遊走によるがん細胞の浸潤には、主に 2 つのパターンが存在します。
浸潤(がん)
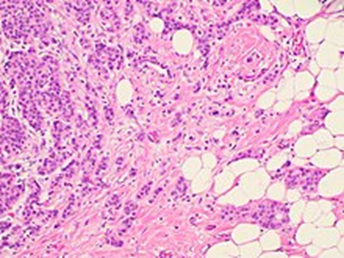
乳房の非特殊タイプの浸潤性癌の組織病理学。画像右側の脂肪組織に不規則な腫瘍巣が存在することから、浸潤性であることが確認されます。
浸潤とは、がん細胞が直接広がり、がん細胞の隣接組織に浸透するプロセスです。一般的に、浸潤は転移とは区別されます。転移とは、循環器系やリンパ系を通じてがん細胞がより離れた場所に広がることです。しかし、リンパ血管浸潤は一般的に転移の最初のステップです。
細胞移動による癌細胞の侵入には、集団細胞移動と個別細胞移動の2 つの主なパターンがあります。個別細胞移動では、腫瘍細胞が細胞外マトリックスの障壁を乗り越えて周囲の組織に広がります。各細胞移動パターンは、異なる形態学的特徴を示し、特定の生化学的および分子遺伝学的メカニズムによって制御されます。
2種類の移動する腫瘍細胞、すなわち間葉系(線維芽細胞様)とアメーバ状は、癌細胞の浸潤のさまざまなパターンで観察されます。この記事では、癌細胞の移動の変種間の重要な違い、上皮間葉系および関連する移行の役割、ならびに腫瘍浸潤におけるさまざまな腫瘍因子と間質分子の重要性について説明します。浸潤パターンの形態学的発現は、さまざまな組織(腫瘍)構造によって特徴付けられます。
浸潤性増殖と転移
悪性腫瘍に関する数多くの実験的および臨床的研究の結果は、浸潤性増殖と転移が腫瘍進行の主な兆候であり、これら2つの密接に関連したプロセスを構成していることを示しています。
悪性腫瘍は、転移カスケードと呼ばれる生物学的現象を引き起こす能力によって定義されます。転移カスケードは、細胞侵入に続いて癌がさらに進行し、離れた臓器や組織に転移が形成されるという複雑な多段階のプロセスです。大規模な転移病変は臓器不全の発症につながります。複雑な侵襲性転移プロセスの「終点」、つまり原発性腫瘍の周辺組織への侵入と転移巣の形成の間には、いくつかの段階(血管内侵入、全身循環内での生存と存在、血管外漏出とそれに続く腫瘍細胞による臓器のコロニー形成、臨床的に検出可能な転移の形成)があり、これらの段階の通過は腫瘍の成長の正常な発達とその後の進行に厳密に不可欠です。腫瘍の成長に伴い周囲の細胞外マトリックス構造への圧力が高まりますが、組織微小環境は腫瘍細胞への圧力を高めることで機能的・解剖学的完全性を維持しようとします。悪性腫瘍の成長を制限する要因には、基底膜と周囲の間質のさまざまな成分、間質圧の上昇、腫瘍細胞への酸素供給の制限と活性酸素種の生成、免疫系細胞との持続的な接触などがあります。腫瘍内の不均一性により、一部の腫瘍細胞は退縮して死滅する可能性がありますが、反対の微小環境因子に対して耐性のある他の腫瘍細胞は、攻撃的な表現型と転移能力を獲得します。
浸潤性腫瘍の成長は、細胞間接着分子の減少または完全な喪失により悪性細胞が腫瘍塊から剥離することで可能になります。これにより、細胞は異常に高い運動性を獲得し、周囲の間質の硬い構造要素を貫通できるようになります。
上皮細胞が細胞極性と細胞間接着を失い、遊走性と浸潤性を獲得して間葉系幹細胞になる過程は、上皮間葉転換(EMT)と呼ばれます。EMTは、胚発生や創傷治癒などのさまざまな生物学的プロセスの正常な特徴です。しかし、転移の観点からは、腫瘍細胞が体の他の領域に浸潤するのを促進します。
侵入性増殖の生理学的プロトタイプ
腫瘍細胞は、さまざまな生理学的プロセスにおいて、腫瘍以外の正常な細胞に典型的に見られるメカニズムと移動パターンを再現する能力を持っています。正常細胞と同様に、腫瘍細胞はこれらのメカニズムを活性化して形状を変え、移動に好ましい条件を作り出し、近くの組織を再形成して移動経路を形成します。しかし、腫瘍細胞は正常細胞とは対照的に、これらのプロセスを停止するための生理学的「停止信号」を持っていません。これが移動メカニズムの確立につながり、腫瘍の進行と拡散を促進します。
悪性細胞は、浸潤性増殖と転移を決定するプロセスを実行するために、組み込まれた遺伝子プログラムを使用していることが判明しました。たとえば、胚発生中および炎症中に観察される個々の細胞の動き(例:白血球の移動)は、腫瘍の進行および転移中の癌細胞の拡散に似ています。
単一細胞の移動に加えて、しっかりと相互接続された腫瘍細胞のグループが一緒に移動すると、集団細胞の移動が発生する可能性があります。このタイプの移動は組織の再配置を示し、胚の形態形成のプロセスの基礎となり、また創傷表面の治癒に不可欠な要素でもあります。
このように、悪性腫瘍細胞は、浸潤増殖および転移の過程において、集団細胞移動と単一細胞移動の両方のメカニズムを生理学的プロトタイプとして利用します。
侵入性増殖のパターン
浸潤性増殖の 2 つの異なるパターンは、特定の形態学的および分子遺伝学的パラメータに基づいて区別されます。単一細胞遊走と集団細胞遊走です。遊走タイプは主に組織微小環境の特性によって影響され、腫瘍細胞内の分子変化に依存します。
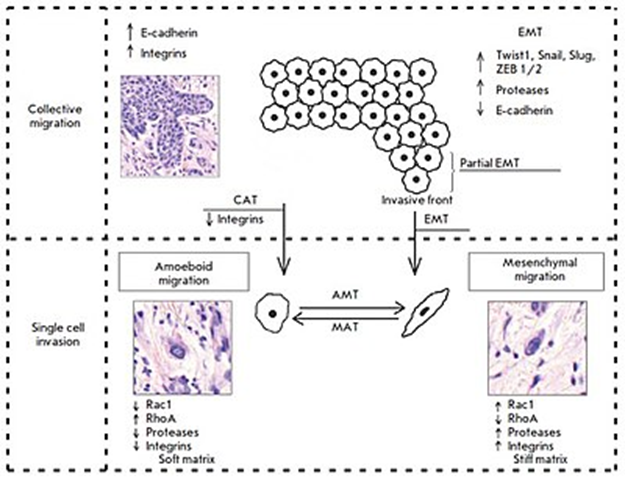
がん細胞の浸潤パターン:集団細胞と個別細胞の移動。集団細胞の移動では、腫瘍細胞はE-カドヘリンとインテグリンの高発現を示します。上皮間葉(EMT)遷移と集団アメーバ(CAT)遷移は、集団細胞浸潤と個別細胞の移動の間の引き金となります。EMTには、TWIST1、Snail、Slug、ZEB1 / 2などの転写因子の活性化、E-カドヘリン発現の低下、プロテアーゼ活性の増加が伴います。EMT中、腫瘍細胞は間葉系の表現型を獲得し、腫瘍塊から分離して、間葉系のメカニズムによって移動します。対照的に、腫瘍浸潤前線に特有の部分的EMTは、腫瘍細胞が細胞間接着を保持しながらも、すでに移動能力を持っていることを意味します。この腫瘍細胞表現型は「上皮間葉系」表現型と名付けられました。β1インテグリンがダウンレギュレーションされると起こるCATでは、腫瘍細胞が腫瘍塊から分離し、アメーバ機構によって移動する。アメーバ状移動には、プロテアーゼとインテグリンの発現の減少、 GTPaseの活性の変化( Rac1の阻害とRhoAの活性化)が伴う。この移動タイプは、緩い/柔らかい細胞外マトリックスで発生する。対照的に、間葉系移動は反対の表現型と関連しており、密で硬いマトリックスで優勢である。これら2つの移動タイプは可塑性が高く、細胞外マトリックスのタイプと細胞内制御に応じて、互いに変換することができる。
移動中の単一細胞が使用する侵入メカニズムの特定は複雑な作業です。2015年以前は、分子レベルおよび形態レベルでこれを調べる研究は少なく、ほとんどが特定の細胞株を使用して試験管内で実施されていました。
しかし、その後、個々の細胞移動や集団移動中の間葉系細胞とアメーバ系細胞の主な違いを決定する腫瘍細胞の分子遺伝学的特徴への関心を示す研究の数が増加しました。
集団移住
集団移動は、接着分子やその他の連絡接合部によって相互接続された細胞群全体の移動を特徴とする。これがこのタイプの侵入の主な特徴であり、その根底にある細胞メカニズムは、主に単一細胞の移動を決定するプロセスと同じである。
集団細胞移動は、乳がん、子宮内膜がん、前立腺がん、大腸がん、大細胞肺がん、横紋筋肉腫、黒色腫、およびほとんどの扁平上皮がんの発生と進行において観察されています。
集団移動の場合、癌細胞は腫瘍塊の一部であるか、または多細胞集団の形で腫瘍塊から分離し、周囲の組織に浸透して、細く短い弦、クラスター、縞、広い領域、および内腔を持つ構造を形成し、腫瘍浸潤に関与する多種多様な構造要素を示します。
集団移動は、カドヘリンと細胞間ギャップ結合によって相互接続された細胞群全体の移動を特徴とする。移動する細胞群には、インテグリンとプロテアーゼを使用する「リーディングエッジ」または「リーディングフロント」がある。リーディングエッジを形成する「リーダー」細胞と、その後ろの「トレーリングエッジ」に位置する「フォロワー」細胞の間では、遺伝子発現と形態に目に見える違いがある。「リーダー」細胞の形状は、間葉系細胞に似ていることが多く、秩序と構造組織があまり目立たないのに対し、「フォロワー」細胞は、細胞間の接触が密で、より密集したロゼット状の管状構造を形成する傾向がある。
集団移動の場合、腫瘍細胞は先端に突起(仮足)を形成し、インテグリンを使用してアクチン細胞骨格と焦点接触を形成し、細胞外マトリックスのタンパク質分解を行い、腫瘍組織の侵入のための空間を作り出し、その過程でアクチン-ミオシン収縮装置を広範囲に関与させて移動を成功させます。
集団的に移動する細胞群の極性の違いは、「リーダー」細胞におけるCXCR4やCXCR7ケモカイン受容体などの表面受容体の発現によるものです。間質細胞によって産生される成長因子とケモカインは拡散勾配を形成し、細胞極性化を誘導します。これらのプロセスにおけるSDF1 (CXCL12)、線維芽細胞増殖因子(FGF)、および形質転換増殖因子 β (TGF-β)などのケモカインの関与は文献で議論されています。
TGF-βの発癌への関与は2つある。乳腺上皮細胞において癌の初期段階で強力な腫瘍抑制因子として働くTGF-βは、発癌性サイトカインとの相互作用を介して腫瘍の発達に影響を与える可能性がある。TGF-βの発現増加は腫瘍の進行と関連しており、乳癌の後期段階でよく観察されている。TGF-βは腫瘍と間質の相互作用の調節因子であり、乳癌における集団細胞移動を促進する。
リーダー細胞は、腎臓の足細胞、1型肺胞細胞、骨格筋細胞、胎盤などで通常の状態で発現している膜貫通糖タンパク質であるポドプラニンを発現することが確立されています。乳がん細胞におけるポドプラニンの発現は、糸状仮足の形成とE-カドヘリン発現の同時保持を伴う細胞の移動と浸潤を誘導します。
集団的に移動する癌細胞は、隣接する間葉系細胞の能力を利用してマトリックスの構造を変更し、再構築し、その「足跡」をたどる可能性があります。in vitro 実験では、培養物に線維芽細胞を導入すると、連鎖の形で下にあるマトリックスへの集団的な腫瘍細胞の移動が誘発されます。このようにして、線維芽細胞は侵入する腫瘍細胞を誘導し、周囲の細胞外マトリックスを、側面に厚いコラーゲン束があり、中央にマトリックスがない経路に改造します。
LIMキナーゼはアクチンを安定化させる酵素で、腫瘍細胞の集団移動に関与している。このタンパク質は、悪性腫瘍細胞に特徴的な構造で周囲の細胞外マトリックスの破壊の原因となる浸潤突起の形成の制御に関与することが知られている。LIMキナーゼの過剰な活性化は乳がんにおいて見られる。LIMキナーゼ遺伝子の発現が抑制された乳がん細胞は、細胞外マトリックスを破壊する能力を失うため、浸潤能力を失う。
単細胞侵入
単一細胞浸潤は、個々の腫瘍細胞が互いに独立して周囲の組織に浸潤することで特徴付けられます。このタイプの腫瘍浸潤では、単一細胞の移動は、間葉系とアメーバ系の 2 つの異なる移動タイプを介して発生します。これらの移動タイプは可塑性が高く、あるタイプの移動から別のタイプの移動に移行できます (間葉系からアメーバ系へ、またはその逆)。これらの移行は通常、腫瘍細胞が微小環境の特殊性に適応しなければならないときに、特定の細胞分子の活動の変化によって発生します。
間葉系(線維芽細胞様)細胞の移動
侵入細胞増殖の間葉系メカニズムは、アメーバ型移動とは対照的に、より複雑なプロセスの発生と、より多くの細胞分子の関与を特徴とする。
このタイプの移動は、修復再生中のケラチノサイト、内皮細胞、平滑筋細胞、線維芽細胞に典型的に見られる。間葉系の動きをする悪性細胞は上皮極性を失い、線維芽細胞に似た細長い紡錘形をとるため、このタイプの侵入は「線維芽細胞様」移動とも呼ばれる。間葉系侵入は、黒色腫、線維肉腫、神経膠芽腫、その他の悪性腫瘍の発症中に検出されている。
腫瘍塊から分離して周囲の組織に侵入する癌細胞のほとんどは、特定の変化を起こし、間葉系細胞に典型的な形態学的特性と表現型を獲得することが知られています。細胞内の新しい分子的および形態学的特徴の出現に関連する悪性上皮細胞のこの変化は、上皮間葉転換(EMT)と呼ばれています。間葉系侵入のメカニズムは、悪性上皮腫瘍の活発な脱分化が起こり、多細胞群が単一の腫瘍細胞に分裂し始め、間葉系表現型を獲得するEMTの結果であると考えられています。
間葉系型の遊走中の腫瘍細胞は、遊走の 5 段階モデルを構成するいくつかの特定の連続ステップを経る。このサイクルには、次の変化が含まれる。1) 細胞極の 1 つに突起が形成。これは、 β1 ファミリーのインテグリンが急速に関与する、小さな GTPase Rac1およびCdc42の制御下でアクチン細胞骨格が収縮することによって生成されるラメリポディアまたはフィロポディアである。2) 細胞外マトリックスと細胞間の接触部位でインテグリン β1 およびβ3が関与する焦点接着の発生。3) インテグリンを介した相互作用に基づく焦点接触の組み立て、および「細胞-マトリックス」界面でのタンパク質分解酵素 (マトリックスメタロプロテアーゼ、セリンおよびスレオニンプロテアーゼ、カテプシン) の活性化により、周囲の細胞外マトリックスが破壊され、リモデリングされる。 4)ミオシン IIを介した制御下でのアクチン細胞骨格の分極の変化、細胞体の収縮の発生、および 5) マトリックス構造に新たに形成された欠陥を通して後縁を移動に向けて「引っ張る」。線維芽細胞のような侵入メカニズムを使用する細胞は、説明した移動ステップに従うため、その移動速度は遅く、約 0.1~μm/分です。
タンパク質分解と組織構造の再構築の可能性は、腫瘍細胞の間葉系運動が、アメーバ状移動と比較して、細胞の形状と核の最小限の変形においてより小さな変化を受けるという事実を説明しています。
間葉系移動では、腫瘍細胞はアメーバ状移動に比べて、その形状の変化や核の変形が小さくなります。これは、間葉系移動ではタンパク質分解や細胞外マトリックスのリモデリングが頻繁に起こるため、細胞は全体的な完全性を保ちながら組織内をより効率的に移動できるためです。一方、アメーバ状移動は、細胞と核の両方の変形度合いが高く、より丸みを帯びた柔軟な細胞形状が特徴です。これは、細胞が組織の狭い空間を通り抜ける必要がある場合によく発生します。
個々の移動中の腫瘍細胞の挙動は、周囲のマトリックスの硬さに依存します。たとえば、周囲のマトリックスが「硬い」(「密」)な状況では、間葉系またはタンパク質分解性の移動モデルが優勢です。密な組織で間葉系メカニズムを使用する単一細胞の高い移動効率は、さまざまなプロテアーゼの分泌によるタンパク質分解と、間質要素との焦点接触を形成する能力によって説明されます。
線維芽細胞様の侵襲性増殖メカニズムの要点は、細胞の両極および細胞と細胞外マトリックス成分間の強力な接着力、インテグリン(β1およびβ3ファミリー)の顕著な発現、タンパク質分解による組織の破壊とそれに続くマトリックス構造の欠陥形成を伴う組織の再構築、および欠陥を通る単一細胞または細胞鎖の移動である。核の変形は最小限であり、細胞移動の速度は遅いことが観察される。
低分子干渉RNAを用いた関連遺伝子の発現抑制に基づき、GTPase Rac1およびCdc42の特異的活性が間葉系浸潤の特徴であることが実証された。GTPase RhoAおよびそのエフェクターであるROCKキナーゼのシグナル活性化を介してGTPase Rac1を抑制すると、腫瘍細胞の間葉系への移動が阻害される。
アメーバ状細胞の移動
アメーバ状の侵襲的増殖機構は、最も原始的であると同時に、単一腫瘍細胞の移動の最も効率的なモードである。そのすべての特徴は、アメーバであるDictyostelium discoideumなどの単細胞生物の行動や動きに似ている。
臨床試験でインテグリンを阻害する抗体やプロテアーゼ阻害剤を使用すると、アメーバ型遊走を伴う腫瘍細胞が出現する。生体内での悪性腫瘍の研究でも同様の結果が得られた。がん治療におけるマトリックスメタロプロテアーゼ阻害剤に基づく薬剤の適用と腫瘍プロセスの進行との関係が確立された。この関係の説明は、アメーバ型遊走が可能な腫瘍細胞が特定されて初めて可能になった。これらのデータは、接着と細胞外マトリックスの破壊を行う主要分子を使用して周囲の組織に拡散する能力が減少または完全に失われた状況下で、腫瘍細胞がアメーバ型侵入メカニズムに転じ、これが唯一かつ最も効果的な遊走モードになることを示している可能性が高い。
このタイプの移動は、循環幹細胞、白血球、および特定の種類の腫瘍細胞で報告されています。Zijlらによると、アメーバ型の浸潤性増殖は、乳がん、リンパ腫、小細胞肺がん、前立腺がん、および黒色腫で観察されています。
アメーバ状移動の場合、悪性腫瘍細胞は円形または楕円形であることが実証されています。アメーバ状細胞は、変形が速く、周囲の細胞外マトリックスの既存の構造に形状を適応させ、圧縮された状態で狭い空間を貫通する特徴があります。移動と再配置は、細胞膜の「ブレブ状」突起の発達を伴う細胞体の連続した高速膨張と収縮のサイクルを通じて実行されます。これらのブレブにより、細胞は微小環境を調査して、さまざまな障害物を迂回するための最適な移動経路を見つけることができ、腫瘍細胞は細胞外マトリックスの狭い隙間を移動することができます。細胞形状の変化は、皮質アクチン細胞骨格によって生成され、これは、小さな GTPase RhoA とそのエフェクターである ROCK キナーゼによって制御されます。このGTPaseは小さなGTP加水分解酵素のスーパーファミリーに属し、そのメンバーはシグナル伝達に関与し、それによって移動中のアクチン細胞骨格の再編成を含む細胞内で起こるさまざまなプロセスの調節に関与するため、アメーバ型の侵入において重要な役割を果たします。
アメーバ状の侵入機構による移動は、細胞の形状だけでなく、核の形状、および他の内部器官に対する核の向きと位置の変化も伴う。核は最大の器官であり、周囲の細胞骨格よりも硬いため、構造タンパク質の広範なネットワークによって機械的にしっかりと安定化されている。このため、その形状は通常、大きな変化を起こさない。しかし、アメーバ状の移動は、周囲のマトリックスのタンパク質分解の欠如を克服するために、核が著しく変形することを特徴とする。腫瘍細胞は狭い空間や孔を通って移動する必要があるため、この場合の核も最大限に圧縮された状態で発生する。白血球のアメーバ状運動と同様に、移動する単一の腫瘍細胞内の核は、先端に向かって前進する。
間葉系の動きとは対照的に、周囲のマトリックスが比較的低い剛性(「柔らかい」マトリックス)を特徴とする場合、アメーバ状または非タンパク質分解性の移動モデルが優勢になります。たとえば、リンパ系および循環系における腫瘍細胞のアメーバ状移動は、柔らかいマトリックス内の移動と見なされます。
Condeelis と Segall は、 MTCとMTLn3という 2 つの異なる腫瘍株を例に、in vitro および in vivo 条件下での細胞移動の特徴をいくつか明らかにしました。転移能が高く、おそらくアメーバ状の浸潤増殖機構によって移動する MTLn3 細胞は、転移能が低い MTC 細胞よりも上皮成長因子受容体(EGFR) の発現レベルが高いという特徴があります。その移動は、周囲のマトリックス内の血管とコラーゲン含有繊維の存在に関連しています。血管への腫瘍細胞の走化性は、EGFR のシグナル伝達経路によって媒介されると考えられています。
アメーバ様の侵襲メカニズムには、いくつかの特徴があります。細胞と周囲のマトリックスとの相互作用が弱く、局所的な接触がないか弱いことが特徴的です。細胞と細胞外基質との接触部位で受容体の急速かつ非局所的な集合を維持する可能性があります。このタイプの侵襲性増殖ではインテグリンは重要ではありません。重要な側面は、細胞とマトリックスの相互作用部位でのタンパク質分解の欠如と、細胞外マトリックスを破壊するタンパク質分解酵素の発現の欠如です。インビトロ研究では、アメーバ様の侵襲性増殖の場合、これらの特性により、腫瘍細胞が培養物中で最高速度 (20 μm/分) で移動できることが実証されています。
アメーバ状間葉系転移と間葉系アメーバ状転移
個々の細胞の侵入時には、ある程度の可塑性と、ある移動タイプから他の移動タイプ(間葉系からアメーバ系へ、またはその逆)への「移行」の可能性がある。これらのイベントは、特定の細胞分子の活動の変化の出現と、組織の微小環境条件への適応の必要性によるものである。
これらの変化は、アメーバ状-間葉系および間葉系-アメーバ状遷移として説明されています。間葉系型の移動を使用する腫瘍細胞は、細胞外マトリックス構造と悪性細胞との相互作用の安定化に直接関与するシグナルおよび機械的経路が弱まる条件下で、特定の方法で変化し、アメーバ状の動きに移行することができます。細胞が間葉系からアメーバ状の侵襲性増殖に遷移する(間葉系-アメーバ状遷移)メカニズムとして、1)プロテアーゼ阻害剤の適用による細胞周囲のタンパク質分解の減少または完全な廃止、2)アンタゴニストによるインテグリン受容体の活性および周囲の間質要素との相互作用の減少、3)小さなGTPase RhoAとそのROCKエフェクターの活性の増加および安定化が説明されています。 S. Bertonらの研究によると、p27タンパク質は細胞運動の制御に重要な役割を果たしている。特に、試験管内条件下でこのタンパク質が欠乏すると、3Dマトリックス内の細胞で間葉系アメーバ転移が誘発される。
間葉系-アメーバ系転換の逆過程であるアメーバ系-間葉系転換の可能性がある。アメーバ系-間葉系転換のメカニズムはおそらく同じ分子基盤に依存しており、説明されている転換の可能性を決定する唯一の信頼できる過程は、小さなGTPaseファミリーのメンバーの活性の不均衡と、RhoA活性に対するRac活性の優位性であるという仮説がある。
集団から個人への移行
腫瘍細胞は、1 つの腫瘍内で集団的にも個別にも同時に移動することができます。この場合、個々の移動から集団的移動への移行は、悪性腫瘍の侵襲性および転移性を高めるための重要なステップです。たとえば、固形塊から分離した乳がん細胞は、リンパ管に侵入する能力を獲得します。現在、個別に移動する腫瘍細胞が生成される上皮間葉系遷移と集団アメーバ状遷移の 2 つのメカニズムが区別されています。次に、後者、特に EMT を経た細胞は、特定の条件下で上皮表現型を獲得し、腫瘍多細胞複合体を形成することができます。この表現型の反転は、「間葉系上皮系遷移」と呼ばれます。
上皮間葉転換
上皮間葉転換は、腫瘍細胞が上皮層から分離して運動性を獲得するメカニズムであり、これは「運動表現型」と呼ばれ、侵襲性の成長と転移を促進します。このプロセスが癌の進行の重要な要因として発達することは、特定の腫瘍株と実験モデルを使用してin vitroで示されましたが、EMT の発達を確立し、 in vivo条件下で腫瘍細胞とその主な特性を特定することは複雑な作業です。
EMT は形態形成の多くのプロセスの基礎となる。通常の状況(胚発生中)では、線維芽細胞から分泌される肝細胞増殖因子(HGF)によって EMT が誘導されると考えられている。HGF は上皮細胞の膜にある特定のc-Met 受容体に結合します。受容体への結合により、アクチンミコフィラメントの重合の強度とアクチンミオシンフィラメントの収縮性を制御する小さな GTPase システムのいくつかのタンパク質(Cdc42、Rac、RhoA、RhoC )を含むシグナル伝達経路が活性化され、ラメリポディア形成の強度とマトリックス付着細胞の張力を決定します。この場合、アクチンミオシン細胞骨格全体の大幅な再配置と E カドヘリン細胞間接触の喪失が起こります。発癌過程において、上皮細胞はEMTと表現型的に類似した形態学的変化を受けるが、関連するHGFリガンドが存在しない状態で進行する。悪性腫瘍におけるこの変化は、さまざまな癌遺伝子の導入によって誘発される可能性がある。変化の過程で、腫瘍細胞は上皮層を離れ、線維芽細胞のように移動することができ、それによって侵入および転移する能力を獲得する。
EMT の過程では、次のような事象が起こります。悪性上皮細胞は、細胞間の密着した接合部の破壊と細胞接着分子 (E カドヘリンやインテグリンなど) の喪失により頂基底極性を失います。細胞のアクチン細胞骨格が変化してリモデリングを受け、ストレスファイバーが形成されます。ストレスファイバーは細胞膜付近の特定の細胞部分に集まり、その後特定の細胞突起が形成され始めます。上皮の基底膜の劣化が起こり、その結果、細胞間接触のない腫瘍細胞が侵襲的な成長と周囲の間質マトリックスへの浸透が可能になり、活発な移動を開始します。
EMT は、腫瘍組織全体にわたって均等に顕著になることはまれであることが判明しました。このプロセスは、上皮細胞から間葉細胞への細胞移行の強度が変化するという特徴があると考えられます。この点で、「部分的 EMT」と表現することができ、これには浸潤前線のほとんどの細胞が関与しています。部分的 EMT は、細胞がすでに正常な移動に必要な特性を獲得しているが、細胞間の接触を維持し続けている状態です。この表現型はハイブリッド「上皮間葉」表現型と呼ばれ、集団移動する腫瘍細胞の特徴に関連付けられました。
Taddeiらは、EMTはTWIST1、Snail、Slug、ZEB1 / 2などの重要な転写因子の活性化に関連するプログラムの誘導によって発症することを示唆している。その結果、強力なカドヘリン結合が破壊され、極性細胞移動が活性化され、さまざまな分泌プロテアーゼによる細胞外マトリックス成分のタンパク質分解が起こり、インテグリン受容体の機能は保持される。乳がん細胞の浸潤性増殖の可能性を決定する転写因子Prrx1の役割は実験的に確立された。
ジンクフィンガードメインを持つZEB1およびZEB2タンパク質はプロモーターに直接結合し、間葉系マーカー遺伝子の発現を誘導し、E-カドヘリンやその他の上皮マーカーの発現を抑制することが示されています。
同様に、SnailとSlugはプロモーターへの直接結合によりEカドヘリン遺伝子の発現を抑制し、デスモプラキンやクローディンなどの上皮タンパク質の産生を抑制し、ビメンチンやマトリックスメタロプロテアーゼの発現を活性化して細胞遊走を増加させる。Sanchez-Tillo率いる研究チームは、転写因子Snailが正常な上皮細胞には存在せず、腫瘍浸潤前線の細胞で検出されることは癌患者の生存率の低さの予測因子とみなせることを発見した。ZEB1/2、Snail、SlugはTGF-β、炎症性サイトカイン、低酸素症によって誘導されると考えられている。
集団-アメーバ遷移
実験データによると、集団からアメーバへの転移の可能性があることが示されています。集団からアメーバへの転移とは、集団的な多細胞集団の形で周囲の組織に侵入する腫瘍塊が、アメーバ運動を利用する単一の移動細胞に解離することです。この現象は、β1ファミリーのインテグリン受容体の阻害剤の適用によって可能になることが示されています。これらの分子は、細胞間接触の形成と、腫瘍細胞と周囲の組織成分との相互作用の両方で重要な役割を果たすためです。
間葉系上皮細胞転換
2015 年現在、間葉系上皮転換 (MET) の根底にあるメカニズムの調査に特化した研究はありません。ただし、そのような現象の可能性は認識されています。この場合、たとえば乳がんや前立腺がんでは、遠隔転移巣の組織構造が原発腫瘍の構造に似ていることが多いと言われています。Friedl と Gilmour によると、これらのデータに基づいていくつかの仮定を立てることができます。第 1 に、浸潤と転移は EMT がなくても発生する可能性があります。第 2 に、腫瘍組織サンプルの通常の病理検査中に単一の播種細胞を検出するのはかなり複雑な作業であるように思われ、EMT 中のこれらの細胞を識別することは不可能です。第 3 に、腫瘍細胞は一時的に EMT メカニズムを使用して血管内侵入し、遠隔臓器や組織に広がり、そこで上皮表現型に戻ります。この変換は、間葉系上皮転換として説明されます。 METは実験的に誘導され、個々に運動する細胞が多細胞複合体を形成したが、生理的条件下でのMETの分子メカニズムは未だ不明である。Nguyenらは、線維芽細胞増殖因子受容体1(FGFR1)の選択的阻害剤PD173074が、 AP-1タンパク質の活性を制御するMAPKシグナル伝達経路を阻害し、METの発生を誘導することを実証した。特定の腫瘍細胞株で行われたPD173074阻害剤の薬剤としての使用の可能性の調査では、腫瘍の増殖、移動能力、浸潤の明確な抑制が明らかになった。この場合、Snailおよびマトリックスメタロプロテアーゼ3、10、12、13遺伝子の発現の減少と、E-カドヘリン遺伝子の発現の増加が観察された。
乳がんを例にした浸潤性増殖型の分類
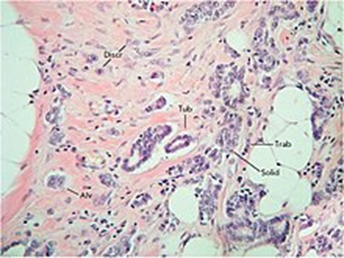
浸潤性乳癌における腫瘍内形態学的不均一性。乳癌の浸潤増殖の多様性が示されており、これは5つの主要な形態学的構造(胞状、小柱状、管状、固形構造、および腫瘍細胞の個別グループ)に分類できます。ヘマトキシリンおよびエオシン染色。倍率200倍。
Krakhmalらは、腫瘍内異質性に応じた乳がんの進行の特徴を研究した。乳がんの組織学的タイプの大部分(80%)を占める、特殊なタイプのない浸潤癌における原発腫瘍の表現型の多様性に注目が集まっている。
原発性乳がんの構造は多様ですが、形態学的構造は主に 5 種類に分類できます。胞状、小柱状、管状、固形構造、および腫瘍細胞の個別のグループです。胞状構造は、円形またはわずかに不規則な形状の腫瘍細胞クラスターです。このタイプの構造を形成する細胞の形態は、中程度の細胞質と円形の核を持つ小型細胞から、不規則な形状と中程度の細胞質を持つ過染色性の核を持つ大型細胞までさまざまです。小柱構造は、小型でむしろ単形性の細胞が 1 列に並んで形成された短い線状の集合体、または中程度の細胞質と円形の正染色性または過染色性の核を持つ中型細胞の 2 列からなる幅広い細胞クラスターです。管状構造は、円形の正染色性の核を持つむしろ単形性の細胞が 1 列または 2 列並んで形成されます。固形構造は様々な大きさや形の領域で、中程度の細胞質と単形核を持つ小さな細胞、または豊富な細胞質と多形核を持つ大きな細胞で構成されています。個別の細胞グループは、さまざまな形態を持つ1〜4個の細胞のクラスターの形で発生します。
乳がんの異なる形態学的構造は、特定のタイプの浸潤に対応しています。したがって、細胞間接触の存在を特徴とする胞状、小柱状、および固形構造は、集団移動の形態学的発現とみなされ、一方、腫瘍細胞の個別のグループは、個別の移動の発現とみなされる可能性があります。細胞接着遺伝子の発現に関する研究で得られた最初の一連のデータは、この仮説を裏付けています。たとえば、細胞間接触を担うカドヘリンの遺伝子の活性は、固形 – 胞状および小柱状構造 – 腫瘍細胞の個別のグループの順に低下しました。この場合、腫瘍細胞の細胞外マトリックスへの接着に関与するインテグリンの発現遺伝子の数は、固形および胞状 – 小柱状構造 – 腫瘍細胞の個別のグループの順に減少しました。
腫瘍の進行と治療効果における浸潤性増殖の種類
浸潤性増殖と薬剤耐性の発達は、腫瘍の進行、特に転移において重要な役割を果たす関連プロセスです。細胞の移動と治療に対する腫瘍耐性の発達には、同じシグナル伝達経路が関与していると考えられます。
移動する腫瘍細胞(移動の種類に関係なく)は、移動しない細胞よりも化学療法や放射線療法に対して耐性があります。これは主に、移動する細胞が一時的に分裂能力を失うという事実によるものです。また、移動する腫瘍細胞は抗アポトーシス遺伝子の活性が増加し、プログラム細胞死の誘導を目的とした化学療法薬に対する耐性を引き起こすという事実によるものです。さらに、EMT状態の細胞は化学療法耐性も示すことが知られています。この薬剤耐性は、EMT中に、化学療法薬を細胞外に排出する役割を担うABCファミリータンパク質の合成が誘導されるためです。EMTを誘発し、同時にABCトランスポーターの活性を正に制御する主な転写因子には、TWIST1、Snailなどがあります。
集団移動と放射線療法および化学療法に対する耐性との間には、潜在的に強い関連性がある。Krakhmal らの研究によると、胞状構造と骨梁構造の両方を含み、かつ顕著な形態的多様性を示す乳がんは、薬剤耐性の増加を特徴とする。骨梁構造が化学療法耐性に寄与する理由は、特定の形態的変異を持つ腫瘍細胞における ABC トランスポーターの活性が高いためと考えられる。対照的に、胞状構造を含む乳がんの耐性は、他の、まだ特定されていない原因によって説明される。
浸潤性増殖およびその表現型の多様性は、直接的にも薬剤耐性の発達を通じても、転移と関連している。将来の転移の発達の原因となる循環腫瘍細胞は、腫瘍細胞がリンパ管または血管に浸潤し、それに続いて侵入した結果生じる。単一の遊走性腫瘍細胞だけでなく、細胞群も血管内侵入能力を持つことができる。集団遊走は個々の遊走と比較して転移につながる頻度がはるかに高いという仮説がある。動物モデルの研究では、単一の腫瘍細胞よりも腫瘍クラスターを静脈内注射した後に転移が形成される頻度が高いことが実証されている。さらに、循環腫瘍細胞クラスターは、さまざまな癌患者の血液中に見つかっている。集団血管内侵入は、VEGF依存性の拡張血管形成および血管内侵入腫瘍クラスターの蓄積に関連していると推測された。さらに、腫瘍細胞の集団は、損傷した血管を介して、または EMT 状態の細胞やプロテアーゼを放出して細胞外マトリックスを破壊する癌関連線維芽細胞との協力によって循環に入る可能性がある。転移は集団移動に依存する。たとえば、閉経後乳癌患者の腫瘍における胞状構造の存在は、リンパ行性転移の高率と関連しているが、閉経前女性におけるこの種の進行のリスクは、異なるタイプの形態学的構造の数が増えるにつれて増加する。後者の依存性も定量的である。乳癌の腫瘍における胞状構造の数が多い場合、リンパ行性転移がより頻繁に検出された。さらに、腫瘍に胞状構造がある患者は、転移のない生存率が低かった(私たち自身の未発表データ)。
集団転移の現れの一つである肺胞構造とリンパ行性転移および血行性転移の速度との関係は、以下の仮説を裏付けている。肺胞構造の細胞要素は、転移表現型を決定する一連の生物学的特性によって、他の構造の腫瘍細胞と異なる。閉経期における肺胞構造とリンパ行性転移の関係は、肺胞構造の腫瘍細胞がリンパ行性経路を通じて転移表現型を獲得するという点で、エストロゲンの特定の役割(その場での産生も含む)を示唆している。
原位置と侵入型
浸潤の程度によって、悪性細胞が腫瘍として存在しているが転移しておらず、発生した層や組織型を超えて浸潤してい ない場合、癌は上皮内癌に分類されます。たとえば、そのような特徴を持つ上皮起源の癌は上皮内癌と呼ばれ、基底膜を超えて浸潤していないと定義されます。対照的に、浸潤癌は基底膜を超えて浸潤しています。これが起こると、癌の浸潤前線はいくつかの分子変化を示し、さらなる浸潤と転移の傾向が増加することを示します。
結論
浸潤性増殖中の腫瘍細胞の移動は、単一細胞を介しても細胞群を介しても起こり得る。この細胞移動の種類の多様性は、おそらく、例えば乳がんにおいて、異なる形態学的構造(胞状、小柱状、および固形構造と、腫瘍細胞の個別のグループ)によって表される腫瘍内不均一性の発生につながる。悪性細胞が周囲の組織に浸潤し、原発腫瘍部位を超えて広がる能力を獲得し、遠隔臓器および組織に二次転移巣の発生を引き起こすことを可能にする、いくつかの生化学的および分子遺伝学的メカニズムが知られている。しかし、異なるタイプの浸潤性細胞増殖と、リンパ行性および血行性転移のパラメータ、疾患進行の特徴、および選択された治療の有効性との間の関係性に関する未調査の疑問が残っている。これらの問題の解決は、疾患の予後を決定し、おそらくは癌患者の管理に対する新しいアプローチを開発するのに役立つ可能性がある。
がんと細胞死「The Hall marks of Cancer」の中で、がん化した細胞の特徴として「細胞増殖シグナルの自己充足」、「細胞増殖抑制シグナルに対する不応答」、「プログラム細胞死(アポトーシス)の回避」、「無限の複製能力」、「持続的な血管新生」、「組織への浸潤と転移」の6つが定義付けられている。私たちの体の中の老化した細胞や傷害を受けた細胞、不要な細胞は死んで除去されるように細胞自身にプログラムされている。しかし、この細胞が死ぬ仕組みに障害が起こると、異常な細胞が増えて「がん」になる。そこで、がんを排除するために、がん細胞に死を誘導することは非常に重要な戦略である。実際、抗がん剤の多くや放射線治療などは、がん細胞に死を誘導することを目的にしている。しかし、既存の抗がん剤に対して抵抗性のあるがんも多く、この試みは現在までに完全には成功していない。さらに、抗がん剤が正常な細胞を傷害することによる副作用も大きな問題である。これらの原因の一つは、生体内で誘導される細胞死の制御メカニズムについての理解が不足していることにある。そこで我々は、生体内において細胞死を制御する仕組みを幅広く研究し、その知見を新たながん治療に応用することを目指している。本項では、がんと細胞死の関わりを概説し、我々が行っている生体内における細胞死の研究と、この研究成果を元にした新たながん治療へのアプローチについて紹介したい。がんとアポトーシスがん細胞は複数の機序によって細胞死を回避することが知られている。もっとも一般的にがん細胞で観察される変化は、がん抑制遺伝子TP53の変異もしくは欠損である。TP53遺伝子にコードされるp53タンパク質は、DNA損傷やがん遺伝子の活性化(発がん性ストレス)などのストレス条件下で活性化され、細胞ストレスセンサーとして機能する。正常なp53は転写因子として働き、細胞周期の停止、アポトーシスの誘導、DNA修復の促進や血管新生の抑制などの重要な細胞機能に関わっている。多くのp53標的遺伝子がアポトーシスの実行に関与していることと、野生型と変異型p53を用いたinvitroの実験において、抗がん剤や放射線照射によって活性化されたp53が標的遺伝子の転写調節領域に結合する能力とアポトーシスを誘導する能力が相関することから、がん抑制においてp53のアポトーシス誘導能は非常に重要であると考えられている。p53の役割はアポトーシスの誘導だけでなく、広く細胞のがん化の抑制に関わっていることから、p53をターゲットにした新たな抗がん剤の開発が精力的に行われている。また、B細胞性リンパ腫において、t(14;18)(:q32;q21)転座によるアポトーシス抑制タンパク質Bcl-2の過剰発現と、それにより引き起こされるアポトーシスの抑制が観察されている。このことから、Bcl-2も抗がん剤の新たな創薬ターゲットになっており、実際に、Bcl-2阻害剤であるVenetoclaxが2016年に米国FDAに承認され、現在、慢性リンパ性白血病、小リンパ球性リンパ腫、急性骨髄性白血病の治療薬として使用されている。これ以外にも、アポトーシスをターゲットとしたがんの分子標的薬として、TRAILなどの細胞死受容体のアゴニストや、IAPの阻害剤などの開発が進められている。IAPの阻害剤については比較的開発が進んでおり期待が持てるが、細胞死受容体のアゴニストについては、invitroの解析では良い成績が出るものの、生体内では奏功していない。シスプラチンやドキソルビシン、エトポシドを含む多くの細胞傷害性抗がん剤や放射線治療もがん細胞にDNA損傷を引き起こすことで、がん細胞にアポトーシスを誘導するが、これらの薬剤はすべてのDNAを標的とするため正常な細胞も傷害してしまい、このことによる副作用も大きな問題である。さらに、アポトーシスの様々な経路に異常が起きているがん細胞に対して、アポトーシスを誘導しようとする戦略は、ある意味で矛盾を抱えている。そこで我々は、生体内において実行される細胞死の制御メカニズムを幅広く研究し、その知見を新たながん治療に応用することを目指している。細胞死研究の新たな展開細胞死の様式には、大きく分けて2つのタイプが存在する。1つは「細胞の自死」とも呼ばれるアポトーシス、もう1つは「細胞壊死」と呼ばれるネクローシスである。図2に示すように、アポトーシスで死に行く細胞は自身に備わったプログラムで小さく断片化し、体内の異物を排除するマクロファージによって除去される。この細胞の死に方は、周囲の細胞に大きな影響を与えずに死んでいくため、静かな細胞死と呼ばれている。一方、ネクローシスは、死の過程で細胞膜が破綻し内容物が周囲に漏れ出る。このため、周囲の細胞に炎症などの影響を与えると考えられている。また、アポトーシスの実行メカニズムの詳細が明らかになる一方で、ネクローシスの制御機構が近年まで明らかにされてこなかったため、「アポトーシス=制御された生理的’図1)アポトーシス経路と創薬ターゲットアポトーシス経路は主にミトコンドリアを介した内因性経路と細胞死受容体を介した外因性経路の2つに分けられる。外因性経路はtumor necrosis factor(TNF)、Fasligand、TNF-related apoptosis-inducingligand(TRAIL)などのリガンド分子が、それぞれの受容体であるTNFreceptor1(TNFR1)、Fas、deathreceptor4/5(DR4/5)に結合することにより、細胞死受容体とFas-associated death domainprotein(FADD)、Pro-Caspase-8が複合体を形成し誘導される。形成された複合体内でPro-Caspase-8が切断/活性化され、下流の実行カスパーゼであるCaspase-3およびCaspase-7を切断/活性化する。その結果、DNaseによるDNAの断片化などを介してアポトーシスが実行される。一方で、内因性経路はDNA損傷などの各種ストレスによって、PumaやNoxaといったBH3-onlyタンパク質を介した、Bax/Bakの活性化によりミトコンドリアからCytochromecが放出されることによって誘導される。細胞内でCaspase-9を含むアポプトソーム複合体を形成し、下流のエフェクター分子であるCaspase-3およびCaspase-7を切断/活性化することにより誘導される。これらのアポトーシスの実行に必要な因子のうち、アポトーシスの外因性経路に関わるFas/CD95、DR5などの細胞死受容体や細胞シグナル因子Fasリガンドをコードする遺伝子、および、ミトコンドリアを介したアポトーシスに関わる遺伝子BAX、PUMA、NOXA、BIDがp53により転写誘導される。さらに、より下流のアポトーシス経路で働くApaf1やCaspase-6をコードする遺伝子もp53によって誘導される。このことから、p53はこれらの遺伝子の転写誘導を通して、発がん性ストレスに対してアポトーシスを誘導し、細胞のがん化を抑制していると考えられる。また、直接的に細胞死に関わる分子に加え、細胞死を抑制するたんぱく質として、チトクロームcの放出に関与するBcl-2ファミリー、Caspasesの抑制に関わるInhibitorofapoptosisprotein(IAP)ファミリーがアポトーシスの制御に重要な分子として存在しており、これらはがんの分子標的として抗がん剤の創薬ターゲットとして注目されている。アポトーシスとネクローシスの一般的な特徴細胞にアポトーシスの刺激が入ると、クロマチンが凝集し、同時に細胞膜の波打ちが観察される。その後、核と細胞質が断片化され、マクロファージなどの食細胞により貪食除去される。この時、細胞小器官の膨潤などは観察されない。一方、ネクローシスの刺激が細胞に入ると、一般に細胞小器官が膨潤し、最終的に細胞膜が破綻して細胞の内容物が周囲に漏出する。45LifeScienceKeyNotesな細胞死」「ネクローシス=制御できない偶発的な細胞死」というイメージが定着してきた。先に述べたHanahanとWeinbergの総説の中でも「programmedcelldeath(Apoptosis)」と記述され、プログラム細胞死(制御された細胞死)とアポトーシスを同義に扱っている。そのため、抗がん剤も主にがん細胞にアポトーシスを誘導することを主眼に開発されている。しかし近年、遺伝子によって制御されたネクローシス型細胞死の存在が広く知られるようになり、その考えが変わり始めている。最近では、アポトーシスとネクローシスといった分類ではなく、熱や物理的損傷などの外傷により引き起こされる細胞死を「偶発的細胞死(AccidentalCellDeath)」、遺伝子によりコードされた細胞死を「制御された細胞死(RegulatedCellDeath)」と呼ぶことが提案されている15。そこで我々は、生理的な条件下の生体内で実行される細胞死について、独自のinvivo細胞死イメージング法を開発して解析を行った。その結果、これまでに明らかにされてきた制御されたネクローシス型細胞死のほとんどが、ウイルス感染や虚血再灌流傷害など病的な場面で観察される一方で、我々はマウス胚発生期の骨形成に関わる新しい生理的なネクローシス型細胞死(Atg9a依存的ネクローシス)を同定した。このAtg9a依存的ネクローシスは、その死細胞周囲に骨形成を誘導するものの、非特異的な炎症は惹起しておらず、何らかの制御機構が働いていると考えられる。このことから、定常状態の生体内においても制御されたネクローシス型の細胞死が実行可能で、さらに、周囲の細胞を炎症等から守る仕組みが備わっていることが示唆された。終わりに紙面の都合でそれぞれの細胞死の実行メカニズムについて詳細を述べることは出来なかったが、近年、これまで制御することが不可能だと思われていた細胞死が制御できるようになってきている。さらに、我々が発見した骨形成に関わるAtg9a依存的ネクローシスのような生理的なネクローシス型細胞死の存在が明らかになったことで、生体内でネクローシス型細胞死を誘導しても、何らかの制御機構によって周囲の細胞を傷害することがないことも明らかになってきた。そこで現在、このようなネクローシス型細胞死の詳細な制御機構を明らかにし、その制御方法を開発することで、既存のアポトーシスをターゲットにした治療法では排除できなかったがん細胞に対する新しい治療法の開発を行っている。この研究はがん細胞に対する新しい武器となるだけでなく、正常な細胞を守る方法にもつながると考えられる。我々の研究グループは、様々な方法を用いて私たちの体の中で起こる細胞死の制御方法について研究を進めており、今後この研究で得られた知見を、がん細胞を排除するための新たな方法へと応用していきたいと考えている。
THINKER logo
がんは光で簡単に殺すことができた
医療従事者でも、ロイヤル・レイモンド・ライフ博士の名を知っている人は、少ないのです。 ロイヤル・レイモンド・ライフ博士ほど、医療において革命的な発明・発見をした人物はいないのです。ライフ博士の偉業は100年前にすべての病気はヘルペスウイルスであることを気が付かないで見つけ出していたのです。
現代のヘルペスが原因であらゆる難病といわれる癌、自己免疫疾患、宿主の遺伝子をヘルペスが簡単に組み換えてしまって生み出した先天的遺伝子病、後天的遺伝子病や原因不明といわれる実はヘルペスが原因であるのです。人々を苦しめ死に至らしめるすべてのあらゆる種類の病気を、完全に治療する方法を、医学も免疫学も今ほど発達していなかった100年も前にすでに見つけていた人物がロイヤル・レイモンド・ライフ博士だったのです。
ライフ博士のすべての病気を治せる治療が本物であることを証明するために私は79歳になってもすべての病気の原因はライフ博士が癌の原因は「癌ウイルス」はヘルペスウイルスであり、しかもワクチンが全く効果がないへルペスウイルスが最後に残された病原体であり、ヘルペスウイルスこそがすべての残された病気であることを理論的、臨床的に完治させることのよって証明するために様々な論文を書き続けているのです。すべての病気を治せる自費診療を79歳になっても臨床で治るという証拠を患者に見せつけるために全世界の病気つくりの医薬業界に虐められながら自分の病気も治すつもりでやり続けています。
みなさん自費診療は高いと思っておられるでしょうが実は国民皆保険こそ一番高い医療なのです。というのは18歳から健康であるときには毎月健康保険料を払い続けているにもかかわらず治らない病気だといわれているにもかかわらず三割負担ですから私の10割負担となる実費医療のほうがが高いと思われるかもしれません。転移癌やがん以外のヘルペスが原因である良性腫瘍や他の難病である自己免疫疾患はすべて完治できるのに治らないといわれて死ぬまで毎月、強制的に健康保険料と三割負担の治療費を死ぬまで払わなければなりませんので、治せる原因医療の松本漢方クリニックのほうが、はるかに安上がりなのです。原因がわからないので他の医者が治せない病気もヘルペスですから私は治せるのです。他の医者が治せない病気の治療にお金を払いしかも対症療法で免疫を抑制し続けてヘルペスウイルスを増やし続ける永遠に治らない現代医療はライフ博士が言うように癌になっていく治療なのですから医薬業界に最後は命を奪われてしまうので無限大の金のかかる医療を安いと思い込まされているだけです。
従って現代の国民皆保険は即廃止すべきで、病気を治したときにのみ病医院は患者からお金を治した報酬としてとれるような医療システムに変えるべきなのです。翻って松本医学は原因医療でありすべての病気の原因であるヘルペスを自分の免疫で細胞内に封じ込める医療ですから免疫を自分で落とし続けない限り必ず感知できる医療ですから世界で一番負担のかからない安い医療なのです。だからこそ老齢にもかかわらず病気を治せる自費診療を続けているのです。
なぜ自費診療にせざるを得なかったのでしょうか?抗ヘルペス剤のアシクロビルを健康保険で病気が治るまで使わせてもらえば国民皆保健医療の中ですべての難病が治ってしまうと病気はすべてなくなってしまいます。病気がなくなると医者がいらなくなるので医薬業界は全滅してしまいます。医師会はそれは困るので抗ヘルペス剤を自費で使えないように「私のヘルペスを自費で使いだすと混合医療という冤罪をなすりつけられてしまいました。自分の病気を含めて現在の治らないといわれる難病の原因はヘルペスであることを知り尽くしている私は保険の中では抗ヘルペス剤を規制が強いので使えないのでどうすべきか苦しみました。しかし何十万人もの患者の診療からすべての病気はヘルペスであることを患者から学び、抗ヘルペス剤と漢方煎じ薬ですべての病気を治していたので仕方なく難病を治すために抗ヘルペス剤を使わざるを得なかったので混合医療を避けるためには自費診療をやらざるを得なくなったのです。ライフ博士のアメリカ医師会の同種のいじめを日本医師会から受けるのと同じ運命をたどっているのですが抗ヘルペス剤を用いることを許されるのは自費診療しか手がなかったのです。世界中の医者で抗ヘルペス剤を合法的に使えるのは私の自費医院しかないのです。
あらゆる難病を治してしまうと医薬業界は破滅するのを避けるために私の松本博士の医学を潰されそうになってしまったのですがロイヤル・レイモンド・ライフ博士の様に4年間も精神病院にも入院しないでかつアルコール中毒にもならず日本の医薬業界の組織的な様々な仕打ちに耐えて、かろうじて自費診療で踏みとどまっています。
15歳から鬱はじめヘルペス性脳炎、ヘルペス性若年性アルツハイマー病など数えればきりのない様々のヘルペス関連疾患やヘルペスによる自己免疫疾患を79歳までの64年間耐えてきたのですがやはり鬱病が自分を苦しめますが自殺する勇気は失ったので何とか生き続けこの世のすべての病気の原因として残されたヘルペス退治に邁進していきます。
二次リンパ器官とリンパ管とは何かを詳しく説明しましょう。
特定の病原体である侵入者に対して特異的な TCR を持つ T 細胞は実は 100 ~ 1,000 個しかなく、これらの T 細胞が活性化されるには、その侵入者を「見た」抗原提示細胞である樹枝状細胞と接触する必要があります。これらの特定の敵だけを認識できる T 細胞と 死の敵を捕まえてくれるAPC は体中に広がって存在しているので、病原体の侵入が完全に制御不能になる前に100 ~ 1,000 個の中の特定のT 細胞とAPCとが出会い起こる可能性は非常に低いのです。この出会いを妥当な確率で機能させるために、免疫系には「T 細胞とAPCとの会合場所」である二次リンパ器官が存在しています。最もよく知られている二次リンパ器官はリンパ節なのです。
家庭には 2 つの配管システムがあります。1 つ目は蛇口から出る水を供給します。これは加圧システムで、圧力はポンプによって供給されています。二つ目はシンク、シャワー、トイレの排水口を含む別の配管システムがあります。この 二つ目のシステムは加圧されていません。水は排水口を流れて下水に流れます。これらの家庭の2 つのシステムは、最終的に廃水がリサイクルされて再利用されるという意味でつながっている2 つの配管システムがあります。
人間の血管とリンパ管の二つの配管はこれによく似ています。一つは心血管系の心臓のポンプによる加圧システム があり、血液はこの心臓のポンプによって体中に送り出されます。しかし、私たちには別の配管システムもあります。リンパ系です。このリンパ系のシステムは加圧されておらず、血管から組織に漏れたリンパ液という体液を毛細リンパ管に吸収して組織からリンパ管に組織からを排出してしまいます。このシステムがなければ、組織は体液であるリンパ液で満たされ、むくみや腫れがひどくなり体全体がむくんでしまい、むくみが増えるばかりとなります。
リンパは上半身ではなく主に下半身の組織から下半身のリンパ管に集められ、筋肉の収縮により、一連の逆流を防ぐために一方向弁を通って上半身へと下半身のリンパ管によって輸送されます。このリンパ(リンパ液)と体の胸より中央寄りの上の上半身の左側からのリンパは胸管に集められ、左鎖骨下静脈の血管に排出されて心臓に戻し、そのリンパは血液と混ざって心臓のポンプ作用によって血管で再び全身に循環されます。同様に、上半身の右側からのリンパは右リンパ管に集められ、右鎖骨下静脈に排出され心臓に戻ります。下の「リンパ管とリンパ節の分布」の図から、組織に出たリンパが血管に再吸収されて血液と再び心臓で合流するために曲がりくねって戻るときに、リンパ管の一連の中継地点である黒い点印のリンパ節を通過することお分かりになるでしょう。
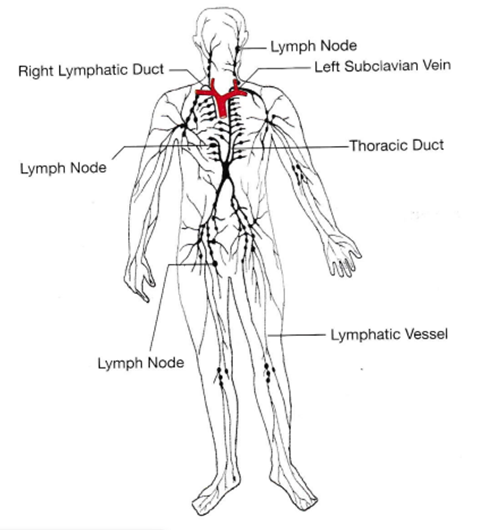
右の図の英語の日本語の訳
Lymph Node リンパ節
Right Lymphatic Duct 右リンパ管
Left Subclavian Vein 左鎖骨下静脈
Thoracic Duct 胸管
Lymph Node リンパ節
Lymphatic Vessel リンパ管
Lymph Node リンパ節
右の絵は「リンパ管とリンパ節の分布」
人間の体には、非常に小さいものから芽キャベツほどの大きさのものまで、約 700個のリンパ節があります。ほとんどのリンパ節は、リンパ管でつながれた「鎖」状に並んでいます。細菌やウイルスなどの侵入者はリンパによって近くのリンパ節に運ばれ、組織内で外来抗原を拾った抗原提示細胞はリンパ節に移動してそのウイルスの積荷を提示します。一方、B 細胞と T 細胞はリンパ節からリンパ節へと循環し、「運命づけられた」自分にピッタリ合う抗原を探します。つまり、リンパ節は実際には「デート バー」として機能し、T 細胞、B 細胞、APC、抗原の四つがすべて集まってコミュニケーションと活性化を行う場所です。細胞と細胞のコミュニケーションとは何でしょうか?多細胞生物である人体の社会秩序を保つためには細胞同士のコミュニケーションが大切です。このコミュニケーションにいつも使われるのはシグナル分子といわれるコミュニケーション専用の化学物質がよく使われます。このシグナル分子を別の細胞が受容体で受け取れば細胞間のコミュニケーションが成立します。
これらの細胞と抗原をリンパ節の小さな容積内に集めると、それらが相互作用して適応免疫系を効率的に活性化する可能性を大幅に高めるためにリンパ節は存在しているのです。リンパ節はあくまでも未熟なT 細胞、未熟なB 細胞、APC、抗原の四つがすべて集まって四つの免疫系が集まってコミュニケーションを行って未熟なT 細胞、未熟なB 細胞の活性化を行う場所です。決して病原体や癌細胞を殺戮するためにあるのではないのです。二次リンパ組織であるリンパ節はいわば教育機関であって戦場ではないのです。それでは何故がんのリンパ節転移が頻繁に起きるのでしょうか?答えは極めて簡単です。強力な兵器や勇猛な戦士がいないのでリンパ節に隠れるためなのです。詳しい説明は後述します。乞うご期待!!!リンパ節の存在価値は初心なT細胞やB細胞を一から教えて大人にする教育機関なのです。教育が終わった後に病原体のいる社会組織に出て戦いを準備する学校に過ぎないのです。だから癌細胞やヘルペスにとっては学校である骨髄やリンパ器官ほど安全な場所はないのです。
リンパ液が流れる管のことをリンパ管と呼びます。組織に出たリンパ液を元のリンパ管に戻し最後は心臓に戻すのです。心臓というポンプから押し出された血液とリンパ液は心臓のポンプ作用で組織に流れ出ていきます。組織に出たリンパ液をそのままにしておくと、リンパ液の量が組織で増えすぎてしまい、むくみや腫れの原因となる可能性があるため、リンパ管が組織に出たリンパ液を血管に戻すことで、正常なバランスを保っているのです。また、血管の血液が体内を円のように循環するのに対し、リンパ液は心臓から血液と一緒に出ていき血液と組織まで運ばれて組織にでていきます。毛細リンパ管の内皮細胞同士がボタン様結合で繋がって特徴的な柏の葉状の輪郭を示し,ボタン同士の間の広く開いた細胞間隙から免疫細胞・脂質・異物・癌細胞などの大型物質を取り込むことができます。リンパ管は末梢組織内で血管と吻合しない網状ネットワークとなっていて,固有の役割を果たすことができる.
リンパ管は体の末端の組織で途切れており、組織に出たリンパ液は自然にリンパ管の内皮細胞間隙から吸収されますが血液の様に循環もすることもできないので心臓で血液と混ざり血液に体内を組織に出るまで循環させてもらうのです。組織にいるウイルスや細菌などの病原体はリンパ管に吸収されて700個以上にも及ぶリンパ節を通過して心臓に戻るのです。心臓に戻る時にリンパ液が逆流しないように弁が付いており、リンパ液は心臓に向かうだけの単一方向にしか流れない点も特徴的です。
組織で病原体を抗原提示細胞である樹枝状細胞を捕まえたこの病原体をB細胞やT細胞に提示して合うB細胞やT細胞を探すために700個以上にも及ぶリンパ節を旅しまわるのです。するのです。つまりリンパ節とはB細胞やT細胞と抗原提示細胞である樹枝状細胞の三つが出会って樹枝状細胞がB細胞やT細胞を活性化するためにデート(密会)するためのバーなのです。つまり直径3センチ位の芽キャベツや豆粒の大豆のような小さいリンパ節が人体の全身に700個もあるのはB細胞やT細胞と抗原提示細胞である樹枝状細胞の三つが集まる目的は自然免疫の樹枝状細胞が適応免疫のB細胞やT細胞と相互作用してB細胞やT細胞を活性化する確率を高めるための教育機関というべきなのです。何故がん細胞はリンパ節に転移したがるのでしょうか?がん細胞はリンパ節で免疫機構により攻撃を受けるといわれていますが本当でしょうか?そもそも癌細胞は人間の遺伝子23500個の中の2種類の癌関連遺伝子は合計で800個近くあります。この2種類の癌関連遺伝子のワンセットの二つが変異起こしてこの2個の異常なタンパク質ができた細胞を抗原提示細胞である樹枝状細胞のTLRが危険なシグナルと認識して敵が来ましたとB細胞やT細胞に伝えて2個の異常なタンパク質を攻撃して炎症が生まれると思いますか?無理です。遺伝的多型とみなし同じ種の中でみられる遺伝的な個性として広くみられる遺伝子のばらつきで作られる蛋白質は異物としてみなすことをしないので病気を起こすことにはならないのです。顔の違いや性格の違いは遺伝子的多型のばらつきで作られる蛋白の違いですから病気ではないのです。美人でないとか頭がよくないというのは病気ではないのです。それでは癌の下人は何ですかに対する答えはヘルペスであるのです癌細胞には非常に多くのヘルペスが増殖しているのです。このヘルペスは病原体のウイルスですから免疫の力が弱いリンパ節にこぞって逃げ隠れするために転移しやすい癌の原発巣の最も近いしかも免疫の弱いしかもヘルペスの怖さを何も知らないナイーブT細胞のいる所属リンパ節に転移し始めるのです。癌についてはロイアルレイモンド博士のここを読んでください。
何故がん細胞はリンパ節に転移したがるのでしょうか?癌では初期からリンパ節転移が観察されるのは何故でしょうか?リンパ組織に侵入する機序は詳しくわかっていないようですので私が説明してあげましょう。免疫のイロハがわかっていないナイーブTリンパ球ばかりが活性化のためにひしめいているリンパ節は全身で700ヶ所あり、そこで癌細胞が転移してもナイーブTリンパ球は癌を殺す力はないので癌細胞は生き残り続けます。リンパ液の流れも圧が掛からないために緩やかでさらにリンパ節の周りには筋肉の収縮もないのでリンパの流れが更に弱くなるので居座ることになり癌細胞の増殖に伴って増殖に専念できます。がん細胞もヘルペスがリンパ節で増殖し固定的な転移巣となりリンパ節の辺縁洞や髄洞にヘルペス性癌を貪食しようとして血管から集まった大食細胞すぎてリンパ節が腫れてくるのです。さらに増えすぎた癌細胞は輸出リンパ管から次のリンパ節まで転移したり、静脈を通じて他の組織に転移することもあるのです。転移した先で増殖し、やがて正常細胞よりもがん細胞が勢いをつけ栄養を得るようになってしまうことで、それぞれの臓器が正常な働きをおこなうことができなくなることもあります。
リンパ節には様々な臓器からの癌転移が見られます。 肺癌の場合、血液やリンパ液の流れに乗って癌が広がるため、リンパの流れが密集しているリンパ節に転移することが多いそうです。 乳癌ではリンパ節に転移する確率が最も高いと言われています。 胃癌の場合は、癌細胞が胃の粘膜を侵食していくことでリンパ節に転移します。
がんは大きく成長すると、一部のがん細胞がリンパ液や血液に乗って他の臓器に転移します。
細胞はなぜ死なないのでしょうか?
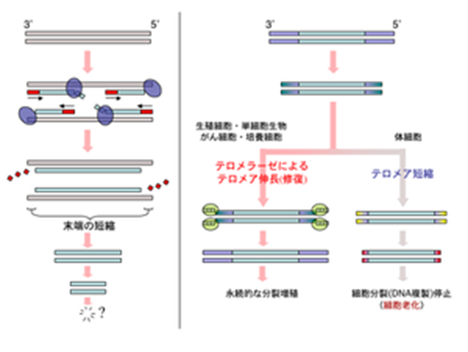
がん細胞ではたいてい、不死化をもたらすテロメラーゼ(telomerase)と呼ばれるテロメア合成酵素が活性化しており、この酵素の働きによってテロメアが安定に維持されます。がん細胞が無限に分裂出来るのはこのためです。
テロメラーゼ(telomerase)とは何でしょうか?
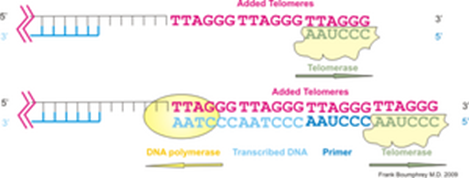
左図はテロメラーゼによるテロメア配列付加の模式図です。ヒトのテロメラーゼは染色体末端DNAの 3’側に6塩基配列 TTAGGGを付加する。左下図は付加された配列をテンプレート(鋳型)としてDNAポリメラーゼが相補鎖を合成する。下左図は末端複製問題とテロメアの図です。DNAはDNAポリメラーゼ(青丸)によって複製されるが、最末端のプライマー(赤線)部分は複製されない。このため、複製のたびにDNAは短縮する。これが「末端複製問題」である。下右図は生殖細胞やがん細胞ではテロメラーゼによって末端部分の複製が行われる図です。テロメラーゼ活性がない体細胞では分裂ごとに短縮がおこり、一定以上短くなると分裂を停止し細胞老化が起こる。
テロメラーゼ (英: telomerase) は、真核生物の染色体末端(テロメア)の特異的反復配列を伸長させる酵素。テロメア伸長のテンプレート(鋳型)となるRNA構成要素と逆転写酵素活性を持つ触媒サブユニットおよびその他の制御サブユニットによって構成されている。
テロメラーゼ活性が低い細胞は、一般に細胞分裂ごとにテロメアの短縮が進み、やがてヘイフリック限界と呼ばれる細胞分裂の停止が起きる。テロメラーゼは、ヒトでは生殖細胞・幹細胞・がん細胞などでの活性が認められ、それらの細胞が分裂を継続できる性質に関与している。このことから、活性を抑制することによるがん治療、および活性を高めることによる細胞分裂寿命の延長、その両面から注目を浴びている。
酵素によりテロメアが伸長されることは、1973年にアレクセイ・オロヴニコフ (Alexey Olovnikov) によって最初に予測された。彼はまた細胞老化に関するテロメア仮説およびがんとテロメアの関連について示唆を行った。
1985年にカリフォルニア大学のキャロル・W・グライダーとエリザベス・H・ブラックバーンは、テトラヒメナからこの酵素を単離したことを公表した。グライダーとブラックバーンはジャック・W・ショスタクと共に、テロメアとテロメラーゼに関する一連の研究で、2009年ノーベル生理学・医学賞を受賞した。
概要
構造と機能
テロメラーゼはテロメア配列の鋳型となるRNAと逆転写酵素、その他の制御サブユニットからなる複合体である。RNA構成要素はTERC (Telomere RNA Component, TRとも表記) 、逆転写酵素はTERT (Telomere Reverse Transcriptase) と呼ばれる。このRNAの長さはテトラヒメナで159塩基長、ヒトで451塩基長、出芽酵母で約1,300塩基長と様々である。逆転写酵素の活性部位はRNA型トランスポゾンがコードするそれと相同性がある。過剰発現の実験から、テロメラーゼ活性自体はRNAと逆転写酵素の二つの構成因子で十分であることがわかっているが、テロメラーゼは生体内において巨大な複合体 (1MDa以上) を形成しており、正常な機能には他の構成サブユニットも必要である。
ヒトのテロメラーゼは、TERT、TERC、ジスケリン (dyskerin) 、TEP1などのサブユニットによって構成されており、それらは異なる染色体上の遺伝子座にコードされている。TERT翻訳産物(タンパク質)は、非翻訳RNAであるTERCと一緒に折りたたまれる。TERTは一本鎖テロメア反復配列を付加できるように染色体の周囲を覆う二股の構造をとる。TERTとテロメアの鋳型を含むTERCは隣接している。ヒトTERCでは鋳型配列領域は 3′-CAAUCCCAAUC-5’であり、これを元にTERTはテロメアの3’側へ塩基を付加する(脊椎動物では6塩基配列5′-TTAGGG-3′(GGTTAG)を付加するが、他の生物では別の配列)。テロメラーゼは、この塩基付加を繰り返し、染色体のテロメアの伸長を行う。
コクヌストモドキ (Tribolium castaneum) TERTのタンパク質構造の詳細な解析が、2008年に行われた。このTERTは4つの保存されたドメイン(TRBD, fingers, palm, thumb)を含むタンパク質であり、レトロウイルスの逆転写酵素・ウイルスのRNAポリメラーゼ・バクテリオファージのDNAポリメラーゼ(ファミリーB)と共通の特徴を持つ環状構造をとっている。
テロメアおよびテロメラーゼの分子機構に関する実験には均一な細胞群を用いることが求められるため、主に出芽酵母やテトラヒメナといった単細胞生物、および哺乳類では培養細胞を用いて研究が行われている。テロメラーゼは細胞周期のS期(DNA合成期)にテロメアに誘導されて機能する。出芽酵母の研究では、テロメラーゼは細胞内で最も短いテロメアから優先的に伸長させていくことがわかりつつあり、長すぎるテロメアには抑制的に働く機構が見いだされている。
活性
テロメラーゼの活性については、生物・組織・細胞の種類によって異なることが知られている。真核単細胞生物は例外なくテロメラーゼ活性を持ち、真核多細胞生物では生殖細胞にはテロメラーゼ活性があるが体細胞での活性はさまざまである。植物においては調べられた殆どの体細胞でテロメラーゼ活性があり、このことが株分けなど栄養生殖でほぼ無限に増殖できる不死性を持つ一因になっていると考えられている。ヒトでは生殖細胞・幹細胞以外での活性がほとんど見られないが、同じ脊椎動物でも魚類・マウス・チンパンジーでは体細胞でのテロメラーゼ活性が観察されている。
ヒトでのテロメラーゼ構成要素の発現をみると、RNA構成要素TERCは体細胞でも発現しており、酵素活性は触媒サブユニットTERTの発現で調節されている。ヒト培養細胞でゲノム中のTERTを強制発現をさせることは困難であるが、人為的に別のプロモーターを付加したTERTを導入することにより細胞の不死化を行うことができる。ヒトのがん組織の多くではテロメラーゼが大量に存在しており、がん細胞の不死化の原因の一つと考えられている(一部のがん組織はテロメラーゼ陰性)。また、生殖細胞は個体を超えて世代を継続させる一種の不死性を持つが、テロメラーゼが恒常的に発現していることがその一因となっている。
臨床関連
がん
ヒトのがん組織の多くではテロメラーゼの活性化がおきており、その観察される割合は肺がんの80%から食道がんの95%に及ぶ。一方、テロメラーゼ活性の制限要因であるTERTの発現が見られない腫瘍も観察されており、それらではALT(Alternative Lengthening of Telomeres)と呼ばれるテロメア長を維持する別の機構が見出されている。
ヒトのがん細胞とテロメラーゼとの関係についての説明は以下のようなものである。
- ヘイフリック限界に達した正常細胞は、p53遺伝子やRb遺伝子などのがん抑制遺伝子の働きによって細胞分裂停止が起きる。
- がん抑制遺伝子に突然変異が起きた細胞は、上記の限界を超えて分裂を続け、テロメア短縮もさらに継続する。
- 通常はこれらの異常細胞は、p53経路による細胞死か、染色体の構造異常による細胞死を迎える。この過程の中で、ゲノムの不安定化が起こり、さらにさまざまな変異を誘発することがある。
- 上記の中の一部には、TERTの恒常的発現を獲得する変異細胞があり、それらはテロメラーゼの活性化を通して染色体を安定化させ、腫瘍形成に至る。
このようにテロメラーゼ遺伝子自体ががん化の直接原因ではないが、がん組織の形成増殖にとってテロメラーゼ活性化が必要である場合が多いため、テロメラーゼを標的とした抗がん剤の開発が行われている。その例として、ヒトTERC(hTR)を標的とするGRN163/GRN163L(ジェロン社)、テロメア短縮を誘導するBIBR1532(ベーリンガーインゲルハイム社)、テロメア構造の安定化に作用するテロメスタチンなどがある。
個体老化
遺伝的な要因による早老症として、ヘリカーゼ遺伝子を原因とするウェルナー症候群・ブルーム症候群 (Bloom syndrome) 、DNA修復に関連するキナーゼ遺伝子ATMを原因とする毛細血管拡張性運動失調症、ゲノム損傷修復の関連遺伝子を原因とするナイミーヘン症候群 (Nijmegen breakage syndrome) などが知られている。これら早老症患者の細胞では分裂寿命が短くなりテロメア短縮も早まる事例があるとする報告や、さまざまな早老症がテロメア短縮と関連しているとする報告がある。これら遺伝子群のDNA修復機能とテロメア長の維持との正確な関連は判明しておらず、個体老化(早老症)とテロメア短縮との関連について研究が進められている。
テロメラーゼを利用した療法が、ヒトの老化を回避して寿命を延長させることにも使われると考える医師もいる。しかしながら、テロメラーゼの活性化には、細胞老化防止の可能性と、正常細胞のがん化の一因となり個体寿命の短縮化をもたらす可能性があることの両面が指摘されており、アンチエイジングへの応用についての評価は定まっていない。
他のヒトの疾患
テロメラーゼおよびその触媒サブユニットTERTは、がん・早老症以外の疾患との関係も調べられている。
TERTと関係すると考えられている疾患としては、再生不良性貧血、猫鳴き症候群 (Cri du chat Syndrome,CdCS) がある。CdCSは、5番染色体短腕(5p)の末端部分の欠損を含んでいる複雑な障害である。TERTは5p領域(染色体上の位置 5p15)にあり、TERTの欠損はCdCSの原因または関与要因として示唆されてきた。
先天性角化異常症 (dyskeratosis congenita, DCまたはDKC) は、テロメラーゼ活性と関連する骨髄の疾患である。症例の35%はX染色体連鎖(伴性遺伝)の劣性遺伝子疾患であり、5%は常染色体性の優性遺伝子疾患、残り60%は原因不明である。X連鎖劣性の場合はジスケリン(染色体上の位置 Xq28)、常染色体性優性の場合はTERC(染色体上の位置 3q26.2)またはTERTの変異が原因となっている。DC患者は、いろいろな他の徴候だけでなく、異常な網状皮膚色素沈着、白板症(口腔粘膜の白い肥厚)と爪の発育異常として現れるひどい骨髄不全を示す。X連鎖または常染色体性どちらのDC患者でも、同じ年齢のほかの人よりも短いテロメアと欠陥のあるテロメラーゼ活性を示す。常染色体優性DCの患者家系の一つでは、世代が進むにつれ、テロメア短縮の率の増加および発症する年齢の低下現象(表現促進)、すなわち各々の世代での悪化が見られる例も存在する。
転移した癌はなぜ治らないのですか?
転移した癌の治療は、初期の癌に比べて難易度が高くなります。これは、転移した癌細胞が体内の複数の部位に分散しているため、全ての癌細胞を除去することが難しいからです。血液やリンパの流れでがんが転移し全身に行き渡ってしまうと、手術でがんを取り除くことは不可能です。もし仮に手術で取り除こうとすると、手術時間が長くなるだけではなく、取り除く量も多くなり、体への負担が大きく、体力、免疫力を奪ってしまい、逆にがんの勢いが増してしまう可能性がありますといわれていますが癌の原因がヘルペスでありかつ転移も上で説明したようにヘルペスが大きくヘルペスが関わっていることを考慮していないのでロイアルレイモンド博士の「癌光療法」で癌ウイルスであるヘルペスウイルスを殺せば転移癌であろうがなかろうが、いかなる臓器のどんな癌であろうが完治できます。
ガン細胞は体温が何度で死滅しますか?
温熱療法とは 腫瘍を電磁波で体外から癌細胞を標的にして42.5度以上加温する治療です。 人間のどんな細胞であろうが42.5度以上に温度があがると死滅します。
癌細胞は何年で癌になる?
1cmの大きさに成長するまでに要する時間は10年から15年といわれています。1つのがん細胞が1センチの大きさになるまで10年から15年かかりますと言われていますが間違いです。しかし、1センチのがんが2センチになるには、1年から2年しかかかりません。
この説明もヘルペスの増殖と癌の増殖との関連は一切考慮されていませんのが問題になり正確さに欠けます。癌の増え方にヘルペスが絡むと何故正確な癌の増殖の数値が出なくなるのでしょうか?それはヘルペスの増殖は癌患者さんの免疫の強さに大きく左右されるからなのです。勿論がん治療の違いによって癌患者の免疫が以外によって大きく影響を受けるので患者の免疫を落とせば確実にヘルペスが増えてしまうので癌細胞の転移も増えかつ癌関連遺伝子の癌化も増えるので癌の増殖と癌転移の拡大も比例してしまうからですが現代の癌学者やがん専門医は誰も気が付いていないので癌に関する数値は全く信用できないのです。いずれにしろ現代の医療のすべては多かれ少なかれ免疫を落とすことになるので「癌は放置するのが一番長生きできる」という故近藤誠先生の意見は正しいのですが彼は癌の原因はヘルペスであるとかヘルペスが免疫低下で増えるので癌も大きくなるなどは一切知りませんので彼は癌を医療で治すことができなかったのです。ロイアルレイモンド博士や松本博士の様に癌ウイルス(ヘルペスウイルス)が癌の原因であると知っている人は世界中に何人いるでしょうか?誰もいないでしょうね。残念です。現在は医者から完全に見放されて緩和医療しかないと診断され見放されている末期がん患者はともかく良性腫瘍や癌ではあるが転移していない悪性腫瘍の患者は治せるという確信をも持ち始めています。だって38年臨床経験で10人以上の癌患者さんが治ったのは抗ヘルペス剤と免疫を上げる大量の漢方洗剤で完治させたからです。癌を治したすべての患者さんにはあなた自身が完治させたのです。癌になったのはあなたがストレスでステロイドホルモンを出しすぎて免疫を抑えすぎたためにヘルペスを増やしたために二つの癌を作る関連遺伝子を癌化させたために癌になったのです。癌を大きくさせたり転移させたくなかったらストレスを減らしなさい。そうすれば癌の原因であるヘルペスが増えないので癌は治る可能性が生まれるよ。私は細胞の中でヘルペスが増えないようにするための抗ヘルペス剤と増えたヘルペスが細胞から出た時に免疫を上げる漢方薬を出すからね。癌を作らないためにも癌が大きくならないためにも更に転移しないためにも免疫が一番大事だからね。といっただけで癌も自分の免疫でしか治せないのです。現在のすべての病気を治すのは自分の免疫しかないのです。今も昔も免疫を最高度に落とすステロイドホルモンがすべての病気の原因であるヘルペスを増やしてしまうのであるので免疫を落として症状をとる対症療法は病気つくりの最高の薬です。
後天性の遺伝子病である癌も先天性の遺伝子病も後天性の遺伝子病も細胞に感染したヘルペスが核のゲノムの染色体に自分のゲノムを組み込みかつ部分特異的組み換えを意図なくたまたま偶然ではあるけれども遺伝子を組み替えたための必然の結果細胞のゲノムのDNAの配列が変わって異常なたんぱく質を作らざるを得なかったので生じた突然変異によって生まれた遺伝子病です。
なぜ癌を治す医療がアメリカ医師会の会長であったフイッシュベインに潰されたのかという誰もが抱く疑問の答えは、医療の正史には決して出てこない、抹殺された天才の壮絶な人生が物語っています。隠され続けた発明と、ライフ博士の生涯を振り返ってみることにしましょう。
ロイヤル・レイモンド・ライフ博士は多くの専門分野を持つ科学者であり医学者であったのです。下に彼が発明した「癌光装置」と彼と奥様の写真を掲載します。

ロイヤル・レイモンド・ライフは、機械系の技術者であった父ロイヤル・レイモンド・シニアと母アイダ・メイ・チェイニ-の次男として、 1888 年 5 月 16 日に米・ネブラスカ州で生まれました。母は、ライフが生まれて 8 ヶ月後に病気で他界しました。この病気はおそらくがんであったと思われます。母親の死の悲しみが彼を癌を完治させる研究に駆り立てたと思われます。その後は、叔母のナイナが、 17 歳までのライフ少年を自分の子どものように育てました。
幼いころから様々なことに興味を持ち、多才で聡明だったライフは、医者になることを決意し、アメリカの最高の医学部のジョンズ・ホプキンス大学に進学し、医学を勉強し始めました。次に細菌学に興味を持ったライフは、ハイデルブルグ大学で多くの菌類標本の写真の撮影に成功しました。その分野での多大な功績をたたえ、後の 1914 年には、同大学から寄生虫学の名誉博士号を授与されています。また、微生物の観察に関し、当時の顕微鏡の倍率の限界に不満を感じたライフ博士は、世界中で随一の技術を有するドイツの光学レンズ会社・ツァイス社で働き、光学レンズについての知識を深めました。

その後、ライフ博士は、病理学における微生物の生態研究を深めるためにカリフォルニアに移ります。そこで、知りあった東洋系アメリカ人女性メイニー・クインと知り合い恋に落ちます。当時の強い人種差別の風潮の中でも、ライフ博士は、彼女に対する思いを変えることはありませんでした。その想いを詩にして手紙に書き、二人は結ばれます。 1912 年に結婚し、子宝には恵まれませんでしたが、メイニーはライフ博士のよき伴侶であり続け、その結びつきは 1957 年にメイニーが亡くなるまで続きました。ライフ博士は1971年、83歳で亡くなりました。
ライフ博士は、専門分野をいくつも独学で身につけており、それまでの科学の枠にとらわれない答えを直感で探し当てることができる科学者でした。本当に優秀な才能に満ち溢れていたライフ博士は、一人で各分野の科学者や技術者が集まったチームのように、知識や技術を自由に操ることができたのです。ゆえに、新たな目的のために新しい技術が必用とされるときに、ライフ博士はすべて自分で機械を発案し、設計していました。ライフ博士の数々の発明品の中には、ヘテロダイン・紫外線顕微鏡やミクロ解像管、極微操作装置などがあり、広範囲にわたる知識を有していたことがわかります。ロイヤル・レイモンド・ライフ博士は1888に生まれ1971年に亡くなりました。ヘテロダインとは何でしょうか?ヘテロダイン(heterodyne)とは、ラジオや信号処理で、2つの振動波形を合成または掛け合わせることで新たな周波数を生成することです。信号の変調および復調、興味のある情報を扱いやすい周波数帯域に移すなどといった用途に使われる。 この操作は真空管やトランジスタなどの信号処理デバイスを使って実施されます。
ロイヤル・レイモンド・ライフ博士が1933 年に発明した驚異の光学顕微鏡「ユニバーサルマイクロスコープ」とはどんな顕微鏡だったのでしょうか?1910 年代当時、すでに知られていた癌やその他の病気の原因である病原菌やウイルスの真の正体をとらえようと研究していたライフ博士は、当時の顕微鏡の性能に限界を感じていました。当時の顕微鏡の倍率の限界であった 2500 倍では、実際に病気を引き起こしているウイルスを見ることはできないと、自らの手でより優れた性能の顕微鏡の開発に乗り出します。光学顕微鏡とは、可視光線および近傍の波長域の光を利用する、顕微鏡の一種。単に顕微鏡と言う場合は、光学顕微鏡を指します。光を用いる光学顕微鏡の場合、一般的に数十倍から1500倍ぐらいまで拡大でき、0.2μmくらいの大きさまで見えます。 ゾウリムシやヒトの卵子、大腸菌といったミクロの世界の観察ができます。また、光の代わりに電子を用いる電子顕微鏡になると100万倍ぐらいまで拡大できますがウイルスや細菌を生きたままで観察できません。電子顕微鏡の見える限界は0.2nm(ナノメートル)です。光学顕微鏡の最高倍率は目視での総合倍率の最大値は,生物顕微鏡で 1000 倍~1500 倍,金属顕微鏡で 1500 倍~2250 倍である。対物レンズが主にその役割を担います。

そして、 1920 年までにライフ博士は、ウイルスを見ることができる世界初の顕微鏡を完成させました。また、 1933 年には、約 6000 個の部品で作り上げた驚くほど複雑な装置である「ユニバーサル・マイクロスコープ」を完成させました。
これは、 60000 倍(6万倍)もの倍率を誇る光学顕微鏡です。 この驚異的な顕微鏡によって、 ライフ博士は生きたままウイルスを観察した世界初の人間となったのです。 60000 倍という拡大率は、今日の技術水準からしても、驚異的な数字といえます。現在でもの光学顕微鏡の最大倍率は2500倍であるからです。
ちなみに、現代の電子顕微鏡を使用すると、その観察下で即座に全ての微生物は死んでしまいます。そのミイラ化した残骸か死骸が観察できるだけなのです。一方、ライフ博士の顕微鏡のもとでは、生きているウイルスが目まぐるしく動き回り、環境の変化によって形を変えたり、発がん性物質と反応して素早く複製したり、また正常な細胞をがん化させていく様子を観察することもできたのです。ライフ博士の顕微鏡の凄さがわかりますか?極小の微生物である癌ウイルスを生きた状態で 観察できるのは、診断と治療という目的のためには、非常に重要なことでした。何故同じものを100年後の現在では簡単に作って癌細胞にいるヘルペスウイルスを観察しないのでしょうか?やはり癌を治してしまうと金の生る木がなくなってしまうからです。悲しいですね。これが人間の性なんです。悲しいです。
上の写真はユニバーサルマイクロスコープライフ博士が設計したユニバーサルマイクロスコープです。1933年に製造されたこの3号機は、6万倍の倍率を持ったウイルスを生きたまま観察できる機能を持っている光学顕微鏡です。
博士の技術はどれ位優れていたのでしょうか?ライフ博士は6万倍という抜群の拡大率を持つ顕微鏡を作るだけでは、無色のウイルスを見るには不十分であることに気付きました。既存のアニリン染料で無色のウイルスを着色することはうまくできなかったのです。ウイルスは染料のコロイド粒子を吸収するには小さすぎたのでした。アニリン染料とはアニリン染料は天然染料ではない合成染料で、そのアニリン染料で染めた革がアニリン染めの革です。しかしその後、化学染料の全般が「アニリン染料」と呼ばれるようになっています。
そこで、 ライフ博士は、まずスリット分光器を使って、辛抱強く様々な病原菌、細菌、ウイルスのひとつひとつの分光学的特徴を特定しました。その後、石英ブロックのプリズムをゆっくりと回転させ、研究対象の微生物に特定の波長の光を当てます。この特定の波長は、現在では立証されている「すべての分子は固有の周波数で振動している」という事実に基づいて、分光学的特徴である固有の周波数と共鳴するものを選択したのです。
すべての分子を形作る原子は、エネルギーの共有結合をした分子構造の中で、その分子固有の電磁波周波数 を出したり、吸収したりしています。そして、同じ電磁波振動やエネルギー特性を持つ分子は二つとしてありません。ちょうど海で二つの波が重なると激しさが増すのと同じように、ある分子とその分子固有の周波数と同じ波長の光が重なると共振現象によって光が増幅され、強まるのです。
共振する特定の色の波長の光を微生物に当ててやると、それまで通常の白い光のもとでは見えなかったその微生物が 、その色の光に共振してあざやかにその姿を現します。ライフ博士はこのようにして、通常の光では見ることのできない微生物を見て、それが活発に細胞組織を侵食していく様子を観察できたのです。
このユニバーサル・マイ クロスコープを使ってライフ博士が見ることのできた微生物のうち 75 %は、紫外線を使ってのみ観察できるものでした。そして、紫外線は可視光線の波長の範囲外の光なので人間の肉眼でみることができません。そこで、ライフ博士は初期のラジオ放送ではよく用いられていたヘテロダイン(周波数を変換する技術)を使い、この問題を解決しました。
それはどのようにするかというと、まずそ の微生物に共振する紫外線の波長のうち、二種類の異なる波長の光を当てます。紫外線であるこれらの光は、重なり合うとお互いの波長を妨害しあい、それぞれの振動数を弱めます。このように二つの光の波が干渉しあい、お互いの波を弱め合い、打ち消しあうことによって、より長い波長の光が新しく生まれます。これは、可視光線の範囲の波長の光であるため、肉眼で見ることができるのです。このようにして、ライフ博士は、現代の電子顕微鏡でもまねのできないウイルスを生きたまま観察する技術を完成させたのです。

1929年11月3日付のサンディエゴ・ユニオン紙。トップ見出しで「地元の科学者が病原菌の驚きの世界を解明」として、ライフ博士と博士が映像に収めた菌類の顕微鏡写真を数点を掲載しています。 左の上部の連続写真は、ストロンギロイデス属の鉤虫が活動する様子をフィルムに収めたもので、その左下写真は破傷風菌(胞子の状態)を217,000倍(21万7千倍)に拡大した世界記録の映像であると注釈があります。1929年11月3日付のサンディエゴ・ユニオン紙。トップ見出しで「地元の科学者が病原菌の驚きの世界を解明」として、ライフ博士と博士が映像に収めた菌類の顕微鏡写真を数点を掲載しています。 上部の連続写真は、ストロンギロイデス属の鉤虫が活動する様子をフィルムに収めたもので、その左下写真は破傷風菌(胞子の状態)を217,000倍(21万7千倍)に拡大した世界記録の映像であると注釈があります。鉤虫とは鉤虫(こうちゅう)は線虫の一種で、腸に線虫感染症を起こします。かゆみを伴う発疹、呼吸症状、および消化管症状を引き起こし、最終的に持続的な失血により鉄欠乏性貧血を生じさせます。破傷風菌とは破傷風(tetanus)は、Clostridium tetani(破傷風菌)が産生する神経毒素(tetanospasmin)による神経疾患である。1889年にエミール・フォン・ベーリングと北里柴三郎が初めて純粋培養に成功した。
ライフ博士の認められなかった偉大な功績はいくつかあります。特筆すべきことに 、ライフ博士は、 なんと 1920 年にはヒトにがんを作るウイルスをすでに発見していました。 正常な細胞をがん化させる研究を 2 万回以上試みたのです。さらにこの微生物の培養液から 400 種類もの腫瘍を作りだすことにも成功しました。そして、これらの過程をすべてフィルムや写真に収め、その詳細にいたるまで記録に残し、このがんウイルスを「クリプトサイズ・プリモーディアルズ」と命名したのです。「クリプトサイズ・プリモーディアルズ」はライフ博士がみつけた癌ウイルスの名前ですが残念ながら公的には認められなかったようです。
癌ウイルスとは染色体のゲノムに癌ウイルスのゲノムに組み込ませて染色体のゲノムの遺伝子を組み替えて遺伝子を突然変異させて2種類の癌関連遺伝子を癌化させることですからこの能力を持っているのはヘルペスウイルスですから「クリプトサイズ・プリモーディアルズ」とはヘルペスウイルスであると断言できます。つまり彼言う癌ウイルスはヘルペスウイルスだったのです。クリプトサイズ・プリモーディアルズを色々調べたのですが彼の研究に関する資料がユダヤ人のアメリカ医師会の会長であったフイッシュベインがすべて燃やし尽くしたので無理でした。
ライフ博士は、 1930 年代当時の科学者には信じられないほどに進んだ境地に達していたため、理解できる人がいませんでした。そのため、多くの科学者は、米・サンディエゴにある博士の研究所を実際に訪れて、自分の目でその事実を確認したのです。

左の写真はバージニア・リビングストン博士ですが彼女は、ニュージャージ 州からライフ博士の研究所のあるサンディエゴに引っ越して、頻繁にライフ博士の研究所を訪れました。彼女は、がんを引き起こす様々な細菌を特定した研究書を 1948 年から出版し始め、今ではその功績が讃えられる有名な科学者です。彼女は、後に研究書の中で、癌ウイルスを“プロジェニター・クリプトサイズ”と新しく名付けています。しかし、ライフ博士の名前は、彼女の研究書の中には、一切記されていません。実際、このようにライフ博士の研究による大発見のほとんどが、ライフ博士の功績としては認められていないのです。バージニア・リビングストン博士の功績は、がんを引き起こすウイルスを特定し、発見したことで医学界で認められていますが、ライフ博士と交流していた事実は医学史において認められていません。
ライフ博士の顕微鏡でウイルスが様々に形を変化する様子を目撃しなかったトーマス・リバーズ博士との激論がありました。当時ライフ博士の顕微鏡でウイルスが様々に形を変化する様子を目撃した科学者とそれを見たことのない科学者との間では、激しい論争が起きていました。当時、影響力のあったトーマス・リバーズ博士などは、何の調査をすることもなく、ウイルスの形態変化を否定します。癌ウイルスであるヘルペスウイルスは子供のビリオンを細胞の中で増やすために形態変化をするのは当たり前のことなのです。細胞の染色体のゲノムに侵入するためにも移動せざるを得ないので生きている癌ウイルスが目まぐるしく動き回り、細胞内の環境の変化によって形を変えたり、発がん性物質と反応して素早く複製したり、また正常な細胞をがん化させていく様子が変化していくのは当然のことなのです。リバーズ博士の劣った顕微鏡では、これらのウイルスの形態変化は観察できなかったため、彼は、「このウイルスの形態変化説というものには論理的根拠がない」と論破したのです。
アーサー・ケンダル博士はライフ博士の研究に協力した科学者です。彼は、微生物が生きたままその中で観察できる培養液( K-medium) を開発し、ライフ博士の研究に協力しました。

現代においても正統派の科学者たちは 、代替医療に関して、まったく同様の観点から評価して結論付けています。前例がないのなら、すべて否定するのです。すべての自己免疫疾患を完治させた松本医学も世界中の医学者から憎まれています。 1930 年代の空の旅は、危険をともなう大変なことであったので、彼らのほとんどは、ライフ博士の研究所があるサンディエゴまでわざわざ足を運んで自分の眼で確認することなどしませんでした。それゆえに、 ウイルスのライフサイクル(生涯過程)は、実際にそれを観察したこともない人たちの手によって結論づけられ、科学的事実として確立されてしまったのです。
ミルバンク・ジョンソン博士もライフ博士の研究を手助けしました。1929年のサンディエゴ・ユニオン紙を見て驚いた友人のアーサー・ケンダル博士から依頼されライフ博士の研究所を訪問、その後、ライフ博士の治療器をがん治療に用いる研究を共にしました。

多くの科学者や医者たちも、暗視野顕微鏡を用いたライフ博士のがんウイルス発見と 、がんウイルスの多様に変化する性質の発見については、その功績を認めるところです。ライフ博士は、当時の最先端をいく科学者たちとも仕事をしていました。
名前を挙げると、大手の非営利医療機関であるメイヨー・クリニック所長を長年務めたE.C.ローズナウ博士、すでに紹介したノースウェスタン・メディカル・スクール理事長アーサー・ケンダル博士、世界的に有名なジョージ・ドック博士、高名な病理学者であるアルビン・フード博士、南カリフォルニア大学学長のルーファス・クレイン・シュミット博士、パラダイスバレー療養所・監督責任者R.T.ヘイマー博士、米国医師会・南カリフォルニア支部長ミルバンク・ジョンソン博士など、その他にも大勢いますがアメリカ医師会の会長であるユダヤ人のフイシュベインは癌を治せるライフ博士の研究を自分のものにできなかったので最悪の敵となってしまいました。経緯は後で述べます。
そのような論争の中でライフ博士は、一切その騒ぎにかかわることなく、ただこの小さな殺人者である癌ウイルスであるヘルペスウイルスを破壊する方法をより洗練されたものに仕上げていく研究に没頭しました。ライフ博士は、その癌ウイルスの姿を視覚化させるのに用いた技術と同じ共振・共鳴の原理を用いてウイルスを破壊することに応用したのです。

左の写真は1931年11月20日にミルバンク・ジョンソン博士が、ライフ博士とアーサー・ケンダル博士の功績を讃えるために催した晩餐会。当時のアメリカのトップの医学博士44名が、「ウイルスを培養し、生きたまま観察できることに成功した」二人を讃えるために集まりました。一番奥に立っている白いスーツの人が、ジョンソン博士、右がケンダル博士、左がライフ博士です。
1931年11月20日にミルバンク・ジョンソン博士が、ライフ博士とアーサー・ケンダル博士の功績を讃えるために催した晩餐会。当時のアメリカのトップの医学博士44名が、「ウイルスを培養し、生きたまま観察できることに成功した」二人を讃えるために集まりました。一番奥に立っている白いスーツの人が、ジョンソン博士、右がケンダル博士、左がライフ博士。
1938年5月6日付のイブニング・トリビューン紙。大見出しで「サンディエゴ在住の科学者、恐怖の病原菌である癌ウイルスは光で破壊できると宣言」とあり、小見出しには「医学界への福音」とあります。またその右には、「ライフ博士、18年間の苦労の末のがん退治」とあります。
ライフ博士は最後に癌ウイルスを破壊する技術も手に入れたのです。ライフ博士は、 すべての物質と同様に、目には見えないレベルでウイルスもそれ独自の固有の振動数で、振動していることに着目しました。そして、そのウイルスと共振する周波数の光を照射して、ウイルスをさらに振動させました。 微生物がその構造的な形を維持するのに耐えきれなくなるまで照射レベルをさらに上げてやると、ウイルスの形は歪み、崩壊してしまうのです。 ライフ博士は、この周波数を「致死反応振動数(MOR)」とよびました。そして、 この「致死反応振動数(MOR)」の光は、ウイルス以外の周りの正常な細胞には、いっさい害を与えないのです。
また、 電気を使用した治療法なので、実質的にわずかな電気代しかかからないため、治療費もたいへん安いものでした。脱毛などの副作用に苦しんだりすることもなく、患者は、すみやかに治療されて、無事に家族の元にもどれるのです。抗がん剤を用いた化学療法や放射線療法や外科手術のように命を危険にさらす必要もありません。
この癌ウイルスを殺す治療法の仕組みは、ワイングラスをある特定の音波で、破壊できることに似ています。目には見えませんが、ワイングラスの分子は、すでにいつも特有の音(その音波のいくつかの高調波と同じ振動数)で振動しています。その音と共鳴しているのです。全てのものは、その物質固有の振動数で振動しているので、その音波で破壊されるのは、そのワイングラスだけです。まさにありとあらゆる全てものは、他とは異なるそれ固有の共鳴振動数を有しており、それこそありとあらゆる周波数が存在しているのです。
この技術を完成させるのにライフ博士は、ヘルペスや小児麻痺、脊髄膜炎、破傷風、インフルエンザなどを含む数多くの危険なウイルスなどの病原菌を破壊する特定の周波数を発見するために一度に丸二日( 48 時間)通しで働くという生活を長年行っていました。
著名な博士たちとの共同研究が始まりました。1929 年、「ライフ博士が開発した顕微鏡でウイルスの生態の観察に成功した」という新聞記事が出ると、アーサー・ケンダル博士が友人のミルバンク・ジョンソン博士にライフ研究所を訪問して、本当か確認してくるよう依頼しました。依頼の通り研究所を訪問した、ミルバンク・ジョンソン博士は、ライフ博士の先進的な研究に興味を持ち、協力者となりました。また、アーサー・ケンダル博士も、ウイルスを培養して、生きたまま観察できる培養液(K- medium) を開発し、ライフ博士と共同研究をはじめました。
そして、 1931 年 11 月 20 日には、ミルバンク・ジョンソン博士による晩餐会が催され、すでに述べたように米国内で最も尊敬される医学界の権威である 44 人が、ライフ博士とアーサー・ケンダル博士の共同研究の功績を祝いました。
1934 年、南カリフォルニア大学は、特別医療研究委員会を設立して、ミルバンク・ジョンソン博士のパサデナ郡立病院の末期がん患者を対象にライフ博士の研究所でがん治療実験を実施しました。その研究チームには医者や病理学者が加わり、患者の診察を行いました。 3 カ月の診療の後、委員会は 86.5 %の末期がん患者が完全に治癒したと報告したのです。さらに治療は継続され、残りの 13.5 %の患者も 4 週間後にはこの治療によって完治してしまいました。ライフ博士の技術による治癒率は何と 100 %だったのです。 現代の最先端治療でさえ、がんの平均治癒率は、 15 ~ 30 %と言われていますから、この数値が、どれほど驚くべきものかおわかりになるでしょう。
しかし、驚いたことに 1939 年までには、晩餐会にまつわる医者や科学者のすべてが、ライフ博士という人物に会ったことなど一度もないと証言する事態にいたります。ともに、研究をしたアーサー・ケンダル博士や、よき協力者であったミルバンク・ジョンソン博士も例外ではありませんでした。いったい何があったというのでしょう。
悲劇のはじまりは1934年にアメリカ医師会会長であったユダヤ人のモーリス・フィッシュベイン氏が、ライフ博士の治療法の独占権を渡すよう要求したのですが、ライフ博士は、それを断ったのをきっかけに始まりました。
悲劇の兆候は、まずライフ博士を買収することから始まりました。 1934 年 には、米国医師会の株式をすべて所有していたユダヤ人のモーリス・フィッシュベイン氏が、弁護士をよこして、ライフ博士の治療法の独占権を渡すよう要求しました。しかし、ライフ博士は、それを断ったのです。
フィッシュベインは 、過去にも、がんの薬草治療を開発したハリー・ホークシー博士を押さえこむために、圧力をかけたことがあります。フィッシュベインは、強力な政治的影響力を行使して、 16 カ月の間にホークシー博士を 125 回も逮捕させたのです。
全ての罪状は、無免許での医療行為であり、裁判では訴追を免れましたが、この度重なる嫌がらせのおかげで、ホークシーは精神的に追い詰められました。ユダヤ人のフィッシュベインは、アメリカ医学協会の会長であり、アメリカ医学協会誌の主任編集員でもありながら、生涯一度も患者を診たことがありませんでした。彼は命を救うことよりも、金銭と権力への飽くなき欲望に意欲をもやす人物でした。
フィッシュベインは、ライフ博士に対して同様の作戦を用いることは裏目に出ると考えたため、ライフ博士は、ホークシー博士のように無免許での医療行為と称して逮捕されませんでした。というのは、もしそのように疑惑をねつ造して、逮捕させてから裁判に持ち込んでも、ライフ博士と研究をともにしていた著名な医療関係者たちが博士を弁護する証言台に立つことになります。そうなると、当然、弁護側は、1934 年のパサデナ郡立病院での臨床試験を持ち出してきます。 医薬品業界が一番恐れているのは、この痛みも費用もかからずに末期がんを 100 %完治させてしまう治療法の存在が明るみに出てしまうことなのです。
フィッシュベインが一番恐れたのは命に関わる癌が完治してしまうとお金が稼げなくなることであったのです。それに付け加え、ライフ博士は長年の研究内容の全てを詳細にいたり、フィルムや写真に収めています。これ以上の明確な証拠はありません。現在そのようなあらゆるがんが治る証拠となる資料がフィッシュベインによって没収されすべて燃やされてしまったのです。何という人類の最大の損失をフィッシュベインがもたらしてしまったのです。
だから、まったく別の方法でライフ博士は潰されたのです。まず、ライフ博士の研究所からフィルムや写真や研究書類の多くが盗まれました。しかし、容疑者が逮捕されることはありませんでした。そして、ライフ博士の研究を立証するため、何億円もかけて設立されたニュージャージー州のバーネット研究所が放火されたのです。
これによって 、ライフ博士も窮地に立たされました。というのは、コンピューターがなかった時代にこれらのデータを復元することは大変なことだからです。さらに、ライフ博士の貴重な顕微鏡は何者かによって、破壊され、 5682 もの部品が盗まれました。そして、最後にとどめを刺したのは、警察による令状なしの捜索と違法な没収です。これにより、 50 年に渡るライフ博士の研究の残骸もすべて処分されてしまったのです。

左の写真のモーリス・フィッシュベインによるライフ博士を亡き者にするための徹底的な圧力と破滅をもたらす陰謀が始まったのです。AMA(米国医師会)の理事長である彼は、医療業界にとって不利益になる数々の治療法を弾圧してきました。
1939 年には、製薬産業を牛耳る一族の代理人は、元ビームレイ・コーポレーション社員の、フィリップ・ホイランド氏を援助して、ビームレイ・コーポレーション社のライフ博士のパートナーを相手取って根拠のない訴訟を起こさせました。このビームレイ・コーポレーション社が博士の治療器を製造していた唯一の会社でした。ホイランド氏は、敗訴しましたが、この訴訟を起こすことによって、ビームレイ・コーポレーション社に莫大な訴訟費用の負担をかけ、倒産に追い込みました。
当時、世界恐慌の時代 のさなか、この会社が倒産することは、ライフ博士の治療器が商業的に生産される道が、完全に閉ざされてしまうことを意味していたのです。
同時に、ライフ博士を擁護した医者たちもすべて、研究費の支給が打ち切られ、職場を追われることになりました。
一方、ライフ博士の治療法について知りながらも、それについて口を固く閉ざした者には、多大な資金的援助があてがわれました。博士の研究を抹殺するには、いっさいお金に糸目をつけなかったのです。なぜかといえば、日本を例にすると、がんの先進医療に対し、一人当たり平均して、約 300 万円(自己負担金額)も費用がかかります。つまり、膨大な利益を生む一大産業なのです。
このような事情から、当時、ライフ博士とがんウイルスの研究を共にしたノースウェスタン・メディカル・スクール理事長のアーサー・ケンダル博士などは、当時では破格の 2500 万円もの恩給を受け取り、さっさと引退してメキシコに引っ越してしまったし、また別の高名な医学博士で、ライフ博士と共同研究したにもかかわらず、固く口を閉ざしたジョージ・ドック博士なども莫大な恩給を受け取り、米国医師会から最高の栄誉ある地位を授与されています。
関係者のすべてが、アメとムチで釣られていく中で、クーチェ博士とミルバンク・ジョンソン博士だけは、ライフ博士の研究を続行することを断念し、もとの処方薬を用いた医療の世界に戻っていきました。
また、製薬企業からの資金で出版され、米国医師会によって牛耳られている医学雑誌は、 ライフ博士の治療について、どんな形であれ掲載することを禁じています。そのため、医学生は大学で勉強中も就職後も、ライフ博士の医学上の大発見について全く知る機会もないのです。
ライフ博士の生きた時代は、まさに文明が急 速に進化していった時です。馬から自動車、そして、飛行機へと。ライフ博士は、 1905 年にアメリカ人の 24 人に 1 人が、がんにかかっていた時代から、亡くなる 1971 年には、それが 3 人に 1 人の割合まで急速に増えていく様をみつめていたのでした。
ライフ博士はまた、 米国がん協会やソーク・ファウンデーションなどその他の多くの医療組織が、彼がサンディエゴの研究所でとうの昔にすでに解決してしまった病気の治療のために、数百億円もの資金を調達し、その後急速に大成長を遂げていった様子もすべて知っていました。 ある時期には、 176,500 種類ものがん治療薬が医薬品として認可されるために検査を受けていたこともあります。これらの中には、わずか 0.17 %でも好ましい結果が得られただけで医薬品として認可されたものもあります。また、致死率が 14 ~ 17 %もあるもので認可されたものもあるのです。
この結果、がんでなく医薬品によって死亡した ケースにおいても、診断書には「完了」とか「部分的緩和」と書かれます。なぜなら、患者は実際がんによって死んではいないからです。事実、 医学界におけるがん治療において重要とされているのは、患者ががんで亡くなる前に薬の作用で殺して、それでがんに勝ったことにしてしまうことなのです。
結論として、ライフ博士の生涯をかけた研究と大発見は無視され、潰されただけでなく、おそらくは、ライフ博士とともに埋もれてしまったものと思われます。ライフ博士の最後の 3 分の 1 の人生は、アルコールに溺れたものでありました。無駄になった 50 年の研究生活からくる精神的な痛み、またすべてを鋭敏に知覚できる意識は、膨大な利益を手にする少数の既得権益者たちの傍らで、無為に苦しむ何百万もの人々を、酒の力なしで、忘れることはできなかったのでしょう。
1971 年、ライフ博士は、バリウム(精神安定剤)とアルコールの過剰摂取により帰らぬ人となりました。 83 歳でした。ライフ博士は、自身の発明した治療器の特殊な光の照射を浴び続けていたため、アルコールの乱用にもかかわらず、これほど長生きできたといわれています。

ビームレイ社の周波数治療器については
左がライフ博士、右側で共振周波数を発するアルゴンガス管を指さしているのは、ホイランド氏。ホイランド氏は同社において、周波数治療器の製造と改良を担当しました。後にAMA(米国医師会)の支援を受け、同社を相手取って長引く訴訟を起こし、ライフ博士を疲弊させました。

バリー・ラインズ著の「The Cure That Worked」(成功したがん治療)という本には、ライフ博士が発明したがん治療とそれを製薬業界・医学界が抹殺し、50年に渡り隠蔽してきた歴史が詳細に書かれてあります。
幸いにも、彼の死とともに彼の電子工学的治療が完全に終わりを告げたのではなく、少数ではありますが、良心的な人道主義の医師や技術者が、ライフ博士の治療器を再現させているようですが本当でしょうか?また、 1986 年には、バリー・ラインズが記した「 The Cancer Cure That Worked (成功したがん治療)」によって、世に知られることになりました。
そして、現在では様々なデザ インや価格でライフ博士の周波数治療器と称する機器が出回っていますが、それがすべてライフ博士の治療と同じ効果を示すかは、わかりません。というのは、ライフ博士の名前を出している治療器のほとんどが、オリジナルのものと、まったく別の構造をしているのみならず最も大切な情報である癌が治ったという情報は一切存在しないからです。あの驚異の100%の治癒率とは、ユニバーサル・マイクロスコープと、ライフ博士の日々の研究の賜物で、はじめて成せる技なのです。ライフ博士の周波数治療器と称する機器が出回っているのは単なる商用目的で、ライフ博士の名前が使用されているため、一般の誤解を生むことにもなっています。
もちろん、このように根源的革新的治療法がはたして実際に存在するのか否かを最終的に判断するには、最終的には癌の原因は癌ウイルスであるヘルペスウイルスが遺伝子を癌化してしまったからです。その癌の原因を殺してしまえば癌を治すことができたのは当たり前のことです。しかもライフ博士は公的に行ったのです。
現代がん医療で使われている医薬品やもろもろの医療行為の研究結果が載せてある公的な医療関係の資料の多くは、すべて粉飾された「二重盲検法」によるものばかりで、その研究は、あらかじめ出資企業の望む結果を出すように仕組まれているのです。というよりもライフ博士の癌の原因は癌ウイルスであると明確に示しその原因を殺してしまえば癌も死んでしまう証拠もあるのですが現代のすべての癌治療は症状が改善するというだけの対症治療に過ぎないのです。
それは、 THINKER 内の健康・医療に関する他のトピックスをご一読いただければ、よくわかると思います。ただひとついえることは、徹底的に圧力をかけてきた歴史、事実があるということです。何の役にもたたない発明には、そんな仕打ちをする必要はないのです。
ライフ博士の周波数治療 は、現在においても、正式な医療としては、禁止されています。この世で一番恐ろしい命を奪い取る癌を治せるのに何故、禁止したのでしょうか?皆さん自分で考えてください。その背景には、かる多くの命があることを忘れてはいけません。ライフ博士を抹殺することで、医療業界の既得権益者にとっては、安泰の日々かもしれませんが、人類が失ったものは、あまりにも大きいのです。
終わり。残念です。他人の苦しみが自分の喜びに感じる人間であることがいやになりますね。
乳癌を中心にして現代の癌治療の疑問点を明らかにしていきます。
がんが原因で死ぬのはどういうことか?とりわけ転移がんで死ぬこと言うことはどういうことか?癌の原因であるherpesの増殖との関わりはどうなっているのか?
癌細胞が増殖して癌が進行していくスピードとヘルペスが細胞の遺伝子を癌化させるスピードと癌細胞が増える進行度合いとはどのようにかかわっているのか?
出産経験のない人や初潮が早かった女性に乳癌の発症が多いのは乳腺が女性ホルモンであるエストロゲン(卵胞ホルモン)にさらされる時間が長いためと言われていますが何故エスエストロゲンはトロゲンを多く作ると乳癌になり易いのでしょうか?エスエストロゲンは代謝されるとコルチコステロンという免疫を下げるステロイドホルモンになるからです。
エストロゲンと乳癌のかかわりは?乳がんのがん細胞の60~70%は、女性ホルモン(エストロゲン)の影響を受けて、分裂・増殖します。 つまり、エストロゲンが乳がん細胞の中にあるエストロゲン受容体と結びつき、がん細胞の増殖を促します。 このように、エストロゲンを取り込んで増えるタイプの乳がんを「ホルモン感受性乳がん」といいます。
女性と男性との癌罹患の割合は?
罹患数の上位5部位(男性は前立腺、大腸、胃、肺、肝、女性は乳房、大腸、肺、胃、子宮)の全がんに占める割合は、男性が6それよりも6.7%、女性が63.6%となっています。
出産経験のない人や初潮が早かった女性に乳癌の発症が多いのは乳腺が女性ホルモンであるエストロゲン(卵胞ホルモン)にさらされる時間が長いためと言われていますが何故エストロゲンを多く作ると乳癌になり易いのでしょうか?答えは後述します。乞うご期待を!!それよりも癌細胞が生まれかつ転移もherpesが組織の細胞同士や細胞と細胞マトリックスが剥がれてがん細胞が転移しない結合は様々な強力な力を持っている接着分子が担っていますが、その強い結合をはがしてしまうのは増殖し続けるヘルペスがもたらすのです。例えば細胞と細胞を接着させる分子に70種類もあるカドヘリンファミリーです。細胞と細胞外マトリックスとの接着に関わるのは細胞内には20種類もあるインテグリンファミリーでありそれらと結合する細胞外マトリック側にはフィブロネクチンやラミニンやコラーゲンがあります。増殖したヘルペスはこれらの接着分子の機能を奪い癌細胞を細胞の塊から切り離してしまうので癌細胞の転移が始まるのです。
つまり細胞の遺伝子を癌化させるのも又一番怖い癌の遠隔転移をさせるのもherpesなのです。癌も転移しなければ死ぬことはないわけですから癌が見つかっても転移を起こすヘルペスを増やさなければ生き続ける可能性が生まれます。従ってヘルペスを増えないようにする治療法にも新たなる光が見えてきました。一番大事なことは免疫を落とさないためにストレスをかけないこと。二つ目は免疫を落とす治療は受けないこと。三つめは免疫を上げる唯一の漢方煎剤を服用すること。四つ目はヘルペスを細胞内で増えないように抗ヘルペス剤を服用することです。言うまでもなく良性腫瘍は癌ではないので今述べた四つの治療法を実行すれば自己免疫疾患と同じく治すことができます。癌転移を如何に防ぐかについては詳しくは後述します。
いずれにしろ癌の原因であるherpesが増えれば増えるほどヘルペスは二種類の癌関連遺伝子が癌化する可能性が増えるのみならずがんが転移する確率も高くなります。現代の三大がん治療は放射線を用いてがんの遺伝子のDNAを傷つけるのみならず正常な細胞のDNAを傷つけて新たなる人為的遺伝子病を生み出しているだけですから意味のない治療となるのです。
放射線ではなくてロイアルレイモンド博士の癌ウイルスの持つ固有の周波数の光を利用する「光癌治療」を行えば副作用も後遺症もなく安全でありかつ癌は完全に治るのになぜ光療法を復活させないのでしょうか?アメリカでは「光癌治療」は禁止されています。癌を治してしまうと医薬業界の「ドル箱」が無くなってしまうからです。資本主義社会は人の命よりもお金の方がはるかに価値があるからです。残念です。ロイアルレイモンド博士の「光癌治療」についてはここを読んでください。癌の原因もherpesであるのに誰も知らないのも不思議です。ロイアルレイモンド博士の「癌ウイルス」は実はヘルペスウイルスであることについてはここを読んでください。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}