そもそも”癌”とは何か?癌はヘルペス感染症にすぎない。しかしこの世で最も恐れるべきはへルペスウイルスであって癌細胞ではないのです。この真実を癌細胞はヘルペスの操り人形であって、癌患者は癌細胞によって殺されるのではなく癌患者を殺す殺人鬼は何とヘルペスであって、癌細胞ではないのです。癌細胞自身はヘルペスの奴隷であって最後はヘルペスを増やしすぎて餓死してしまうのです。癌細胞はヘルペスに操られた人形に過ぎないのです。一番恐ろしいのはヘルペスが癌細胞をすべて支配していることを知らない間違った治療をやり続けて「癌は死ぬ病気」と言いまくって無知な大衆を脅かしている癌専門医たちなのです。
癌の原因はヘルペスであるから癌は理論的には完治できるのです。この真実を詳しく論証していきましょう。更に、癌細胞は見かけ上、永遠に分裂を続けて死ぬことはないとされているのは何故かも明らかにします。
癌は傷ついたDNAの長年の蓄積によって癌関連遺伝子の突然変異で起こったわけではないのです。癌関連遺伝子の突然変異を起こして癌を生み出したのはヘルペスウイルスなのです。「癌関連遺伝子の突然変異」という表現も「分裂・増殖関連遺伝子のヘルペスによる変異」と言い換えるべきです。
何故でしょうか?突然変異率はタンパク質をコードするエキソン部分では極めて低いのです。エキソン以外のイントロンやエキソンを含んでいる遺伝子を除いた部分では極めて高いのです。というのはエキソン内での変化(変異)は有害なことが多くて淘汰(selection)されてしまうのでその変化(変異)は蓄積されることはないのです。ところが癌学者は遺伝子ではないDNA損傷が遺伝子を突然変異させて癌生まれると間違った理屈をこね回して嘘をついてDNA損傷の蓄積が突然変癌ができると愚かな理論を主張しているのです。
実は遺伝子となるエキソン以外の癌に関りのない部分では、突然変異率はほぼ一定で1塩基の変異率は1年あたり5~7×10-9なのです。つまり100万年たつと200塩基のうちの1塩基だけが変異(突然変異)を起こすだけなのです。
仮にエキソンのなかで突然変異が起こり、これが有害ではなくむしろ生存に有益な効果があればこの変異は保存されていくのです。このような有益な突然変異の効果こそが保存されて霊長類で最高の種である「ヒト種」である私たちが進化して生まれたのに遺伝子と直接か変わらないつまりがんを作ることが出来ないがらくたのゲノムでも100万年たつと200塩基のうちの1塩基だけが変異(突然変異)を起こすだけなのに人類を滅亡させる癌が100年も生きられない人にたった10年で奇跡的に癌関連遺伝子に極めて証拠のない原因で突然変異が起こりその変異が積み重なって10年で「癌」が起こることは絶対にないのです。その有害な偽りの「奇跡というべき出来事」が二人に一人に起こりしかも二人に一人が死んでいくのです。癌学者の言うこのような有害な「奇跡」は絶対に絶対にあり得ない理屈なのです。必ず他に癌関連遺伝子を変異させる原因が潜んでいるのです。それがワクチンが作れない現代最後に残ったヘルペスウイルスなのです。
100年前にその本当の癌の原因を発見したのがロイアルレイモンドライフ博士なのです。誰でもが感染してあらゆる細胞のゲノムに潜伏感染して癌関連遺伝子(細胞分裂連遺伝子)を変異・癌化させ癌の原因となった癌ウイルスであるヘルペスウイルスを極めて簡単に「光療法」で殺すことによってすべての転移癌をたった3か月で治すことが出来たのです。ロイアルレイモンド博士についてはここを読んでください。
遺伝子となるエキソン以外の部分でさえ、実は「突然変異率はほぼ一定で1塩基の変異率は1年あたり5~7×10-9」なので、言い換えると100万年たつと200塩基のうちの1塩基だけが変異(突然変異)を起こすという真実を皆さんしっかり覚えておいてください。因みに、100万年たつと200塩基のうちの1塩基だけが変異(突然変異)を起こすという計算を使うと進化の上で二つの種がどれほど前に分岐したかも推定できるのです。この方法で作られたのが進化の系統樹(phylogenetic tree)なのです。
20世紀の間、医学者は癌増殖を促進する細胞および分子のメカニズムを学びました。過去50年にみられる生物医学的研究の加速で、予測可能な方法で細胞機能を変更することが可能となり、疾患の検出および革新的な研究方法が科学界および医学界に提供され、がんなどの重篤な病状を治療するための新しい方法を見つけることが可能となりました。しかしこれらの研究は癌完治を目指す治療のためには全く無駄でした。何故ならば癌を生み出す原因は、つまり正常な細胞の癌原遺伝子とがん抑制遺伝子を突然変異させて癌細胞に変えてしまうのも癌を増殖させるのも癌を転移させるのも最後は癌患者が悪液質で死ぬのもすべてヘルペスのなせる業であるのを誰も気が付いていないからです。この間違いだらけの過去の論文を俎板に載せてひとつずつ無駄な病気作りの研究論文の間違いを免疫の理論を駆使して論理的に批判していきます。癌はワクチンが絶対に作れないヘルペスウイルスが生み出した病気であり遺伝子に感染したヘルペス性感染症に過ぎないのです。癌は本来死をもたらす病ではないのです。医薬業界がとりわけ医者たちがお金儲けのために恐ろしい絶対に治らない病気であると間違った理屈で捏造し無知な大衆に喧伝しまくって作り上げた最高にお金が儲かるヘルペス感染症にすぎないのです。
そもそも癌は特別な病気ではなくワクチンが作れない特別なウイルスであるヘルペスが生み出した後天的な細胞分裂・増殖に関連する遺伝子変異病なのです。癌関連遺伝子を変えて後天的遺伝子病である癌をも生み出する特殊なヘルペスによる感染症にすぎないことを証明していきます。癌が完治できるかどうかは患者の癌細胞の数で決まるのではなくヘルペスが800以上もある細胞の癌原遺伝子と癌抑制遺伝子を突然変異させたかによって決まります。突然変異させた癌原遺伝子と癌抑制遺伝子が少なければ少ないほど早く治るのです。従ってヘルペスを増やさない治療法が癌を完治できこの世から癌を撲滅できるのです。この最高の方法の一つがロイアルレイモンドライフ博士の「光癌療法」です。ライフ博士の「光癌療法」はここを読んでください。ヘルペスウイルスは短い核酸である2重鎖直線状のDNAがキャプシドといわれる保護膜である外皮で包まれています。宿主である人間の細胞の生化学的機構を用いて自分のDNAやタンパク質合成のために細胞が死ぬまで利用し尽くして最後は遺伝子まで変異させて癌細胞を無限大に生み出して悪液質にして人をも殺してしてしまうのです。従って癌細胞自身は何も癌の原因ではなく癌に仕立て上げたヘルペスウイルスだけを恐れるべきなのです。
この論文では、はじめに、癌関連遺伝子が一番多い「がん増殖シグナル伝達」の遺伝子の研究を可能としてきたいくつかの重要なマイルストーン(一里塚)ついて言及されますが癌の根絶には何の役にも立ちません。しかし癌を治せない敵の間違った理論を勉強することは「ヘルペスが癌を作る」根拠をより一層理解しやすくなります。次に、がん研究の進歩において可能となった、癌標的治療の実用化の具体的な方法を図解入りで紹介されます。
癌標的治療とは何でしょうか?分子標的薬とは、がん細胞に特有の分子や遺伝子を標的として、がん細胞の異常な増殖や分裂を抑えることを目的とした治療薬です。がん細胞を殺しても癌の原因であるヘルペスウイルスを殺しているのではないので大金となる治療代の無駄使いだけでなく副作用で苦しむことになります。
分子標的薬は、肺がん、乳がん、大腸がん、胃がん、腎臓がん、血液がん、肝臓がんなどの薬物療法に導入されており、一時的な対症療法で効果を上げているといわれていますが癌を治す薬ではありません。手術後に再発予防の薬物療法として使われる場合もありますが無駄に無駄を積み重ねるだけです。どんな病気も原因療法をやらない限り絶対に病気を治すことはできません。
分子標的薬の副作用としては、皮膚障害、心毒性、間質性肺炎、腸管穿孔、動脈血栓症などがあります。皮膚障害では、皮膚にぶつぶつができたり、爪の周りに炎症ができたり、皮膚が固くなったり腫れたりするものが比較的多くみられます。命にかかわる副作用は起きることもあります。癌分子標的治療とも呼ばれる癌標的治療は,がん遺伝子により産生されるタンパク質などを標的として,その働きを抑えたり,「がん周囲の環境を整える因子」といわれるわけもわからない分子を標的にして,がん細胞が増殖しにくい環境を整える治療法です。がん細胞を増殖させるのはヘルペスウイルスですからこの研究も治療法もすべて無駄です。何故ならば癌細胞を増殖させるのはヘルペスであるからです。「がん周囲の環境を整える因子」にヘルペスは全く考慮されていないからです。又癌の発生や進行に直接的な役割を果たす癌関連遺伝子を「ドライバー遺伝子」と呼びます。ヘルペスによるドライバー遺伝子に変異によって生まれる癌では,ドライバー遺伝子を標的とした薬(分子標的治療薬)が有効ですといわれるのも癌を治すためには全く無駄なことです。異常なドライバー遺伝子に変えたのもヘルペスです。というのは癌細胞の癌化した癌関連遺伝子を標的にする限り癌細胞が増殖分裂するたびにherpesはその何百倍も増えていきます。その癌細胞が2倍になって癌遺伝子を持った癌娘細胞を一個増やすとこの新しい癌細胞の800個もある癌関連遺伝子をherpesが更に癌化させる確率を増やすのみならず更に増えたヘルペスは近隣の多くの大量の正常な細胞にも感染して癌関連遺伝子を新たに癌化させる確率をも増やし癌細胞が否が応でも増やしていくので標的を間違えた癌標的治療で高価な人工の抗体を作ってお遊びで作られた人工抗体で癌細胞を殺しても金が儲かるだけですが正常な細胞も殺してしまうので故近藤誠さんが言うように副作用ばかり多くて金儲けのために製薬メーカーは作っているだけです。癌標的治療敵は本能寺に隠れている最悪の敵はヘルペスなのに研究者は何十年かかっても気が付かないのです。因みに癌を治す研究は150年以上行われていますがすべて的外れの薬ばかりです。ヘルペスを世界中の大製薬メーカーが人工的に協力して作れば協力してやれば極めて簡単に作れるのに作ってしまえば癌をはじめとするあらゆる難病も雲散霧消してしまう医薬業界消えてしまうことになるので誰もやりません。医薬業界がアコギな世界である続けざるを得ないのは病気がなくなれば医薬業界は破滅せざるを得ないからです。
癌細胞が悪性度を増すというのはどんな意味を持つのでしょうか?癌細胞の数が増える以上に一個の癌細胞のみならず近隣の正常な細胞の癌関連遺伝子がどれだけ多くヘルペスウイルスによって遺伝子の突然変異を起こしているかで決まるのに誰も気が付いていないのです。しかも細胞の癌関連遺伝子を癌化させているのは活性酸素でも放射能でも紫外線でもないのです。確かに活性酸素は細胞のDNAを時に障害を与えることはあるでしょうがDNAの障害が即癌関連遺伝子が癌化することにはならないどころかエクソンだけを含む遺伝子を構成するDNAは仮に障害されてもエキソン内での変化は有害なことが多く起こると普段から常にこうした影響を受けていてもエキソン内でのDNAに突然変異が奇跡的にしかど起きないのは、すべての生物が、このようなDNA損傷の脅威に対抗する仕組みである「DNA修復機構」を持っていて、エキソン内での変化は有害なことが多く起こるのでDNAを常に正常に保つために淘汰(selection)されてしまうからです。一方エキソン外のイントロンや遺伝子以外の変異は生存に無関係なものが多いので変異がそのまま蓄積されて行くのです。癌はエキソン内の遺伝子の変異ですから癌のよう様な有害なことが多く起こるのでDNAを常に正常に保つために淘汰(selection)されてしまうので癌をおこすDNAの変異は蓄積されないので現代の癌学者の理論は完全に間違っているのです。
すべてのDNAをゲノムといいますが、ゲノムは決して不変なものではなく常に突然で変化しているのです。変化の度合いはゲノムの部位によって異なります。突然変異率はタンパク質をコードしているエキソン部分では低く、イントロンや遺伝子以外の部分では高いのです。エキソン(エクソン)とは何で、イントロンとは何でしょうか?エキソンとは、真核生物の遺伝子において、タンパク質の合成に関わるDNA配列部分です。真核生物の遺伝子では、タンパク質の情報に相当する部分が分断されている場合がほとんどで、遺伝情報がコードされている部分をエクソン、コードされていない部分をイントロンといいます。エクソンは、最終的に成熟mRNAに含まれ、タンパク質合成の鋳型となります。エクソンは実際に表現されるDNA配列という意味で命名されており、その多くはタンパク質に翻訳されます。真核生物の遺伝子発現において、エクソンはスプライシング反応を経てエクソンだけが選択されます。転写された直後のmRNAは前駆mRNAと呼ばれ、前駆mRNAにはイントロンとエキソンという2つの領域があります。前駆mRNA とはスプライシングなどのさまざまな加工を受けて成熟した mRNA になる前の RNA 分子です。タンパク質をコードする遺伝子の多くがイントロンを持つ。これらの遺伝子は細胞核内でRNAに転写され一次転写産物となる。これはヘテロ核 RNA (hnRNA) とも呼ばれる。その後①5′ 末端へメチル化されたグアノシンが付加する5’キャップ、②RNAスプライシングによるイントロンの除去、③3′ 末端に多数が数珠状に連なったアデニル酸が付加するポリA尾部といった装飾を経て、成熟mRNAとなる。このようにして出来た mRNA は核外に輸送されタンパク質に翻訳されます。
突然変異率はエキソン(エクソン)外の部分ではほぼ一定であり、1個の塩基の変異率は1年当たり5×10-9~7×10-9です。つまり100万年たつと200塩基の内のたったの1塩基だけが変異を起こすだけです。この関係式を使うと進化の上で二つの種が前に分岐したかが推定できます。この方法を使って進化の系統樹が作られたのです。系統樹とは、生物の進化の道筋を描いた図である。生物同士の類縁関係と、それらの系統発生の順序を示す図です。樹木のような形になることから系統樹と名づけられた。
エキソンの中で突然変異が起こり、これが癌のように有害ではなくむしろ有益な効果があればこの変化(変異)は保存されるのです。このような変化こそが進化に関係する突然変異なのです。
淘汰(selection)とはなんでしょうか?選択(selection)とは、進化において、生物個体や形質などが世代を経ることによってその数や集団内での割合を増していくことであり、逆に、割合を減少させていくことを淘汰(とうた)というのです。このような変化が実際に起こることを選択が働く(選択される)、または淘汰が働く(淘汰される)といい、この差を生む要因を選択圧または淘汰圧という。英語ではselectionで、選択のほうが直訳に近い。淘汰とも訳される。選択と淘汰は表裏一体であるのです。そのためこの二つのメカニズムを総称して選択(または淘汰)という場合もある。ただし文脈によっては選択と淘汰を区別しなければならないこともある。選択と淘汰を区別せず、割合を増していくことを正の選択または正の淘汰、割合を減らしていくことを負の選択または負の淘汰と呼ぶこともある。「塩基除去修復」や「ヌクレオチド除去修復」をはじめとする、さまざまな修復系が常にDNA損傷を認識し、除去・修復しているのは淘汰が働いているのです。変異は長期的に見ると進化の一部ともいえるため、完全に変異を起こさないようにしてしまうと進化しなくなってしまうことになります。ある程度の変異が起きつつ、死なない程度の変異があるのですが癌は命を奪い取るのでDNAの損傷を減らす淘汰が働いているのです。生物はこのようにしてDNAの完全性を維持しているのです。いうまでもなくDNAの完璧な不完全性が維持されて癌が生まれると主張しているのが阿呆な世界中の癌学者なのです。何のために?自分だけの快楽を最大限増やすための金儲けのために他人の命を犠牲にして!!!!悲しい悲しい人間性の発露ですね。さらに完全な皮肉を言わせてもらうとそもそも活性酸素を産生するのは酸素ですから「癌になるから酸素を吸うーな!」と毎日毎日教育すべきではないでしょうか?アッハッハ!!!
それではヘルペスは何故、核の染色体の中にあるゲノムに自分のゲノムを組み込んで隠れて、細胞が分裂するまでかつ免疫が落ちるまで潜伏感染をし続けるのでしょうか?細胞が自爆して自分も死んでしまうのを避けるためです。ヘルペスが細胞に感染するとその細胞は何故、又どの様に自爆するのでしょうか?ヘルペスはまず細胞の細胞質に感染すると感染した細胞はヘルペスがゲノムに隠れて癌を含めてあらゆる遺伝子病を起こす恐ろしい敵であることを長い進化のなかで知っているのでヘルペスが自分の仲間である他の細胞に感染しないようにインターフェロンαやインターフェロンβを出して自分の細胞もろともherpesも自爆テロで殺し自殺してしまうのです。このように他の仲間の細胞にヘルペスが感染し続けるのを避けるためにヘルペスが細胞に感染するとインターフェロンαやインターフェロンβを出して自殺するのです。このように人間の細胞は人間よりも他人である細胞に対して優しいことを知ってください。この世に4~5億年前に生まれた恐ろしいヘルペスを自爆テロで細胞は自殺することを学んだのです。ところがヘルペスも賢い敵ですから自爆テロで殺されるのは嫌ですから細胞質にいる限り細胞の作るインターフェロンαやインターフェロンβで死んでしまうのでインターフェロンαやインターフェロンβから逃れるために一番安全な核のゲノムに細胞分裂があるまで潜伏感染することを身に着けたのです。
潜伏感染する仕方の一つは核のゲノムに自分のゲノムを組み込み部分的特異的組み換えをやって隠れることともう一つはエピソームであります。細胞分裂が起こるまで潜伏感染するのは自分の娘細胞を生んで増やすことが細胞分裂ですから流石ヘルペスといえどもこの時はインターフェロンαやインターフェロンβは作ることはしないのです。エピソーム(episome)とは、細胞質内に存在し、染色体外と染色体内の両方の存在形態をとることができるDNAの総称です。エピソームとは細胞が本来もっている染色体とは別に,比較的短い環状DNAが独立した染色体として安定的に維持されたプラスミッドと言われる形ですべてのヘルペスウイルスは自然宿主である人間の細胞の核に潜伏感染を成立させるやりかたです。
プラスミッドとはウイルスが人や細菌の細胞に感染して細胞のゲノムDNAとは独立に複製、分離する小型DNAの総称で、環状の分子が多いのですが、線状のものも知られており、遺伝子組み換え技術の発展と共にくみベクターとして多用されています。例えばヘルペスウイルスの一つであるEBヘルペスウイルスはBリンパ球に感染してバーキットリンパ腫という悪性リンパ腫を起こすときにBリンパ球に潜伏感染する時のEBウイルスの場合,B細胞の分裂に伴ってエピソームは娘細胞に分配されるのです。この時に、Bリンパ球の塩基配列を変えるのでBリンパ球の癌関連遺伝子が突然変異を起こし悪性リンパ腫であるバーキットリンパ腫が生まれるのです。
ベクターとは、外来遺伝物質を別の細胞に人為的に運ぶために利用されるDNAまたはRNA分子である。人間の細胞に感染して細胞のゲノムに侵入したヘルペスウイルスはランダムに自分の遺伝子やDNA配列や、RNA配列を感染先の導入先の細胞質やDNA内で増幅・維持・導入してしまい、いわゆる遺伝子組換えを行って癌関連遺伝子のみならず他の遺伝子を突然変異させてしまって先天的遺伝子病や、後天的遺伝子病を生み出すのです。具体的には、プラスミドやコスミド、ラムダファージ、および人工染色体等を指す。
ラムダファージとはファージ(phage)は、人ではなく細菌や古細菌に感染して複製するウイルスで、正式にはバクテリオファージ(bacterio‐phage)と呼ばれる。ファージの基本構造は、タンパク質の外殻と遺伝情報を担う核酸 (主に二本鎖DNA) からなる大きさは10〜100nm(ナノメートル)ので人に感染するヘルペスは大きさは150nm(ナノメートル)よりもかなり小さいのです。ファージが感染した細菌は細胞膜を破壊される溶菌という現象を起こし、死細胞を残さないのはヘルペスウイルス感染の溶解感染と同じです。細菌が食べ尽くされるかのように死滅するため、これにちなんで「細菌(bacteria)を食べるもの(ギリシア語のphagos)」を表す「バクテリオファージ(bacteriophage)」という名がつけられたのです。
コスミドとは、DNAのクローニングに用いられる特別なベクターで、λバクテリオファージの包装に必要なDNAの部分を含むDNA断片です。ゲノムDNAを約3万から4万塩基対程度の長さのDNAとしてクローン化する目的で使用されます。付着末端配列を組み込んだプラスミドで、コスミドには、λファージのcos遺伝子が含まれているので、「cos+plasmid」という意味です。
「がん」は、重要な細胞機能を制御不能とする特定の遺伝子変異を特徴とする疾患群を表す一般的用語です。がんは、ヘルペスウイルスが体内のあらゆるすべての細胞や器官や臓器に感染するのであらゆる細胞を癌化させてしまい、例えばEBウイルスの様に胃癌などの固形腫瘤を形成するか、あるいは悪性リンパ腫であるバーキットリンパ腫のような血液系のリンパ球を侵す腫瘍を形成します。核を持たない細胞以外のあらゆる核を持つ細胞を癌化させます。
ヘルペスウイルスは癌を作って最後は宿主を殺すために生まれたのでしょうか?違います。20万年まえにミミトコンドリア・イブによって人類が誕生したときは人口も極めて少なく、当然ストレスも少なかったのですが数千年前に4大文明が生まれてから被支配と支配の人間関係が生まれかつ農耕社会が生まれて土地や富の私有化が始まり、所有欲も生まれ、満たされない欲望が無限大に増えてしまい、独占したい欲望を満たすことができなくなるにつれてストレスも無限大になり不満の多い人は耐えるためにステロイドホルモンを出さざるを得なくなり必然的に免疫が落ちて殺し切れないヘルペスが無限大に増えてしまいヘルペス性のまやかしの自己免疫疾患や癌が増えていったのです。エジプトの王のお墓の屍骸にも癌が見つかるほどに王様の地位もいつも脅かされていた証拠です。
「がん」を示す別の用語は別に「悪性腫瘍」および「新生物」があります。全てのがんは、起源の組織に関わらず、細胞増殖の加速、解剖異常細胞の存在、体内の他の組織に転移または浸潤するという共通の特徴を示すのは増えすぎたヘルペスがあらゆる細胞に感染して癌関連遺伝子を癌化させるからです。解剖異常細胞の存在は数多くのヘルペスが核のへの無に組み込んで部分的特異的組み換えを癌現遺伝子に全く関わりのない他の細胞小器官などの蛋白の設計図である遺伝子も組み替えてしまうと解剖学的に異常と観察される細胞が生まれるのは当然のことなのです。
転移性癌はがん末期に生じ、がん罹患者の主要な死亡原因となっているのですがどのようにしてヘルペスウイルスが転移性癌を起こすのかはここを読んでください。転移性癌は癌末期に生じという表現は理解に苦しみます。とくに癌末期の意味が正確に定義されていないからです。癌末期は癌の病状がひどくなってもう救いようがない時期の意味に用いられているのでしょうが何を基準にして命が終わるのかの確実な基準がないからです。癌で近い未来を暗示しているのでしょうが、癌は原因も浸潤も転移もヘルペスが起こしている訳ですから治せる病気ですから「癌は特別な病気で絶対に治らない」という言葉は医者が最も好む金科玉条の根拠のない信念ですから私が最も嫌う言葉です。いずれ癌も完治できる病気であることが一般の人にも知られることになるでしょう。実は今までにすでに大腸がんや悪性リンパ腫などの癌を抗ヘルペス剤を用いて治しているので私の癌完治の理論が正しいことはすでに経験していることなのです。癌完治も何も難しいことではないのです。世界保健機関によれば、がんは世界的に主要な死亡原因で、2020年にはおよそ 1000 万人が亡くなっており、癌死は死亡 6 件のうち 1 件の割合となっています。
医学界では、全てのがんの5%~10%が遺伝性であると認識されています。遺伝性の癌についてはここを読んでください。遺伝性であるのは妊娠中に胎児の2種類の癌関連遺伝子のひとつが癌化していたという意味です。残りの90%~95%のがんは、毒素(タバコなど)、UV照射および他の環境要因、不健康な食事や過剰なアルコール摂取などの生活要因を始めとするさまざまな原因によって生じる遺伝子変異によって発生し、多くのさらなる要因によって、がんが発現するリスクが高まると言われていますが間違いです。知らぬ間に増えすぎたヘルペスウイルスが原因です。人間関係と生活苦と過当競争のストレスを耐えすぎてステロイドホルモンを出し過ぎ免疫を抑制しすぎて乳幼児に両親からもらったherpesが徐々に徐々に知らぬ間に年老いた時に人の一個の細胞に800個もある2種類の癌関連遺伝子が数多くの細胞で癌化してしまいの最後は見てわかる、触ってわかる症状で気が付く癌と認識されるようになるのです。具体的には一個の癌細胞は10年たてば10億個に増え、大きさ1センチで、重さ1グラムなると言われていますがこの計算は大間違いです。現代の癌学者は次のように癌細胞が増えていくと考えています。つまり一個の細胞は必ず一年に3回分裂して10年間で30回分裂するので、分裂するごとに、2倍、2倍と増えていくので、10年で230となるので約10億となる計算です。この計算は根本的に間違っています。元々がんが増殖するのは800個もある癌関連遺伝子(ドライバー遺伝子)の変異の数で決まるものです。その変異の数が確定しない限り確実な末期がんの状態であるかどうかを判断できないのです。しかも未来を確実に予言するにはさらに今ある癌関連遺伝子(ドライバー遺伝子)の変異の数が増える勢いは知る必要がありますが癌細胞がこれから先何回分裂するかとか免疫低下がどれだけ続くかどうかも知ることができない限り余命いくらかであるかは正確に知ることはできません。
何故癌関連遺伝子をドライバー遺伝子と言われるのでしょうか?ドライバー遺伝子とは、がん遺伝子・がん抑制遺伝子といった、がんの発生・進展において直接的に重要な役割を果たす遺伝子をドライバー遺伝子と呼ぶ。又がんの発生過程においては、ゲノム変異が起こりやすい状態(いわゆるゲノム不安定性)となるため、がんの発生には無関係な遺伝子にもランダムに変異が起こることがあり背景変異、あるいはパッセンジャー遺伝子と呼ばれます。ドライバー遺伝子もパッセンジャー遺伝子も曖昧な定義ですから使わないほうが良いのです。というのはすでにドライバー遺伝子に相当する癌原遺伝子とパッセンジャー遺伝子に相当する癌抑制遺伝子があるからです。しかもヘルペスがこれらの遺伝子を変異させることがわかっているので癌原遺伝子は正常な時に幹細胞の分裂・増殖遺伝子として働き、癌抑制遺伝子は幹細胞の分裂・増殖が終われば分裂・増殖抑制遺伝子となるだけですから二つをまとめて癌関連遺伝子といわないでヘルペス性幹細胞制御異常症に変えるべきです。ちょうど精神分裂症という恐ろしい響きのある病名を統合失調症と変えて病気のイメージが用にまともになったように「癌」という言葉は全面的に廃止すべきです。ですからどちらも癌というわけのわからない古代から存在していた「癌」という言葉がなくなることによってこの世に存在しない病気も実は4億年前に単純な出現したヘルペス感染症に過ぎないことが一般大衆に理解されて「癌」を恐れる必要がなくなってしまうでしょう。しかし医薬業界が医薬業界は一番儲かる病気がなくなってしまうと医薬業界も崩壊してしまうでしょう。100年まえに癌を「光癌治療」によって簡単に完治させたロイアルレイモンドライフ博士の様に私の癌完治理論と癌治療も潰されてしまうでしょう。実は「自己免疫疾患はない理論と自己免疫疾患の原因はヘルペスである従って抗ヘルペス剤で完治した臨床」をつぶされたように今度は「癌もヘルペスが遺伝子を変異させた病気に過ぎない。」という理論とかつ臨床もロイアルレイモンドライフ博士と同じように潰されてしまうでしょうか?日本にはロイアルレイモンドライフ博士をつぶしたユダヤ人のフイシュベインはいないのですが集団としてお金のためには組織犯罪も辞さない医薬業界が組織がある限り存在する限り覚悟はしています。アッハッハ!!!!
何故この世にあり得ない自己免疫疾患や癌を生み出すヘルペスが増え続けるのでしょうか?資本主義という人間社会にあふれかえっている生まれつきの能力を無視したお金儲けの過当競争から生まれる膨大なストレスです。言い換えると自分の思い通りにならない社会制度の中から生まれる欲望が達成されない社会的人間的関係です。
毎日生き続けることから生まれる嫌な人間関係と嫌な生活苦と嫌な過当競争のストレスによって免疫が落ちてヘルペスが変わるのは癌関連遺伝子(ドライバー遺伝子)の変異が2種類のドライバー遺伝子が最初の一個の癌細胞を産生するのであって、タバコなど、UV照射(紫外線)および他の環境要因、不健康な食事や過剰なアルコール摂取は一般的な普通の生活の中でたとえDNAが損傷されても直接癌の原因となる癌関連遺伝子が絶対に変異することは絶対にないことはすでに証明しました。ただし慢性的に続く生活要因がストレスが強すぎる嫌な人間関係と嫌な生活苦と嫌な過当競争などの解決不能な長すぎたストレスであれば確実に老化が原因ではないのですが年とれば癌になる可能性は極めて高くなるでしょう。
他のがん関連リスクファクターとして、ヘルペスウイルスと比べて極めて少ない子宮頸がんを起こす癌ヒトパピローマウイルスがあります。胃がんは細菌のHelico‐bacter‐pylori(ピロリ菌)による感染によって起こることを見つけた学者にノーベル賞が与えられましたが私は大いに疑問を感じています。というのはピロリ菌は胃の上皮細胞の二つの癌関連遺伝子を癌化することはできないからです。何故ならばピロリ菌は細胞内に侵入することはできないから核にある遺伝子を突然変異させることができないからです。ところが4番目のヘルぺスウイルスであるEBウイルスが胃の上皮細胞に腺癌を起こすことが判明しました。EBウイルスはピロリ菌と違ってヘルペスウイルスですから遺伝子を変えることができるから胃の上皮細胞に腺癌を生み出すことができるのです。
その機序はDNA 塩基そのものの変化ではなく、遺伝子の転写を司るプロモーター領域のDNA 塩基配列の遺伝子が、メチル化という修飾(-CH3基の付加)を受けているのです。DNA 塩基配列の変化を伴わずにDNA を修飾して遺伝子の発現に関与するしくみをエピジェネティクスと言います。エピジェネティクスな癌とは二つの癌関連遺伝子によっておこる癌ではないのです。エピジェネティクスとは何でしょうか?後成学または後成遺伝学と訳し、一般的には「DNA塩基配列の変化を伴わない細胞分裂後も継承される遺伝子発現あるいは細胞表現型の変化を研究する学問領域です。」EB ウイルスによる胃がんではエピジェネティクスの異常が高頻度、広範囲に起きているのです。プロモーター領域の重要な部分が広く修飾を受けると転写が阻害され、遺伝子の発現が低下します。がんを抑える働きをもつ遺伝子が転写されなくなると細胞の癌化につながります。やはり胃がんもherpesウイルスのEBウイルスが原因であったのです。EBウイルス(エプスタイン・バール・ウイルス)とは、ヘルペスウイルス科に属するウイルスの一種で、日本ではよくEBウイルスと略して呼称されます。学名はヒトヘルペスウイルス4型と変更されましたが、今なお旧称であるEBウイルスが広く用いられています。 EBウイルスは、いわゆる「キス病」と言われる伝染性単核球症の原因ウイルスとして有名である。口唇粘膜から感染しやすい病気なのです。
EBウイルスは直線状二本鎖のDNAウイルスであるが,両端に繰り返し配列(terminal repeat)を持っている.感染細胞の核内で,ウイルスDNAは両端で結合して円環状構造(プラスミド)となるため,個々の感染細胞ごとにウイルス末端繰り返し配列の個数が異なってくるのです。このため,繰り返し配列部位を制限酵素で切り出し,その長さを検索するとEBウイルスのクローナリティーを明らかにすることができます。
クロナリティーとは何ですか?
2個体の生殖により生まれる子孫ではなく、1個体から分裂、複製により増殖した子孫の集団をクローンと呼ぶ。 生命体あるいは生物の構成要素の集団・集合体が、いくつのクローンで構成されているのかを示すのがクローナリティです。すなわちクローナリティとは、その複製集団の起源となる個体の数・量を示しているのです。EBウイルス関連胃癌ではリンパ腫などと同様,症例ごとに6kb以上の単一のバンドが検出されることから,EBVは単クローンであり,潜伏感染の状態であることが証明されるのです。
ウイルス感染症としてEBウイルス関連胃癌をみると,胃癌細胞ではウイルス粒子を産生しない潜伏感染の状態にあります。しかも,EBウイルスによるBurkittリンパ腫と同様,Ⅰ型潜伏感染と呼ばれる状態にあり,EBウイルス蛋白のうちEBウイルス特異核内抗原1(EBV determined nuclear antigen 1, EBNA1), Latency membrane protein 2A(LMP2A)など,ごく少数の蛋白のみが発現しています。リンパ球不死化,線維芽細胞の形質転換に各々関連するEBNA2,LMP1の発現は認められないのです。このため,EBウイルスの発癌機構解明のためには,ウイルスと感染細胞の相互作用,感染細胞自身の異常についても分析する必要があるのです。Burkittリンパ腫とはc-myc遺伝子と免疫グロブリン遺伝子の相互転座がヘルペスウイルスによりc-myc遺伝子が過剰に発現し、細胞分裂を異常亢進させることによるのです。バーキットリンパ腫(Burkitt lymphoma:BL)はリンパ節の胚中心(germinal center)の B細胞を起源とする極めてアグレッシブな成熟B細胞腫瘍であり,骨髄や中枢神経などのリンパ節外浸潤を生じやすい特徴を持つ。遺伝子の相互転座とは異なる2本の染色体に切断が起こり、その切断された断片が交換され、他方に結合するものです。2本の染色体が交差した時に起こりやすいです。流産や不妊が起こりやすくなります。相互転座は、通常は情報が入っている染色体の遺伝子の位置が換わっただけなので、見た目や性質である表現型は変わりありません。このような人は均衡(きんこう)型転座保因者と呼ばれます。保因者は、見た目には何も異常がありませんが、子孫を残すために精子や卵子を作るときに、遺伝情報が過不足した不均衡染色体をもった精子や卵子ができる可能性があります。
EBV 潜伏遺伝子の機能と発がんとの関りについてはまずEBVの潜伏,細胞の分裂のときに起こる再活性化,不死化やがん化の性状はEBV遺伝子発現に依存します。EBVは2本鎖直鎖DNAを持ち,ウイルス粒子内では直鎖状,細胞内では環状絵ピゾームDNAとして,稀に細胞染色体に組込まれて存在する。EBVのDNAには約100遺伝子がコードされ,EBV潜伏細胞では,極めて限られた遺伝子のみが発現している。8種類のヘルペスウイルスのDNAには平均、約100遺伝子から150個の遺伝子がコードされています。EBVencoded -smallRNA(EBER)という特徴的なRNAが発現されている。蛋白としては,核抗原(EBV‐nuclear-antigen略してEBNA)蛋白と潜伏感染膜蛋白(latent-infection-membrane-protein略してLMP)があります。核抗原蛋白には核抗原1,核抗原2,核抗原3A,核抗原3B,核抗原3C,核抗原LPの6個の核抗原蛋白があります。潜伏感染膜蛋白には潜伏感染膜蛋白1,潜伏感染膜蛋白2A,潜伏感染膜蛋白2Bの3個があります。これらの発現様式は宿主細胞の種類によって異なり,これらの蛋白の発現が最も少ない潜伏感染をⅠ型,それより蛋白の発現が多い潜伏感染をⅡ型、及び全ての蛋白が発現されている潜伏感染がⅢ型に分類され,発癌と密接に関っているのです。何故ならばEBVは潜伏感染中に細胞の癌関連遺伝子二つを突然変異を起こして癌を産生するからです。ここで注意をしておきたいのはこれらの抗原蛋白の原料や酵素やタンパや質やエネルギーのすべてを細胞から盗み取っているのです。しかもEBVは細胞が一回分裂するときに何千個も子供(ビリオン)を生み出すので細胞も疲弊しきって栄養不足と酸素不足となり増えすぎたEBVは癌患者を悪液質にさせてしまうのは、いくら大量の新しい血管を作り出しても間に合わず最後は極度の栄養不良となり悪液質で癌患者もガリガリになってやせ衰えて死んでしまうのです。この血管新生も癌細胞のためや他の正常な細胞のために作っているのではなくヘルペスの増殖のためにヘルペスに強制的に作らされているのです。しかも正常細胞も癌細胞も自分たちが分裂・増殖するためのDNAを複製するための大量の塩基もアミノ酸もタンパク質も糖分もリン酸もあらゆる栄養素もすべてヘルペスウイルスに奪われてしまうのでまともな細胞を作れないので癌細胞の顔つきが悪いとか細胞内の小器官も細胞の形もすべて異常になって異形成の程度が高度であると言われるのです。
このような疲弊していく癌細胞なんかは何も怖くないのです。ヘルペスの恐ろしさと癌細胞の怖さを比べれば天と地の違いがあります。当然でしょう。だってヘルペスがいなければガンを含めて自己免疫疾患などの難病のすべてがこの世から消えていくからです。癌そのものは全く怖い敵ではないのです。世界中の人に感染してすべての難病の原因となるヘルペスを免疫を抑え続け、増やし続ける医薬業界が一番恐ろしい冷酷な人間の集まりです。悲しいですね。

左表にアメリカにおける致死性の癌の順位が示されています。圧倒的な第一位の死因は肺がんで年間に159万がなくなっています。二位は肺がんの死亡者数の半分である肝臓がんであり三位が胃がんであり第四位は結腸直腸がんでした。第五位の乳がんでは52万人がなくなり第六位は食道がんの40万人です。なぜ肺癌が癌死のトップになっているのでしょうか?その理由の一つがあらゆる臓器の血流は肺を通ります。血流で運ばれてきたあらゆる臓器の癌細胞はヘルペスを乗せて肺に血行性転移で運ばれます。その理由の二つ目は二種類の癌関連遺伝子がほかの組織の臓器よりもはるかに多いからです。二種類の癌関連遺伝子が多いのは肺には多くの種類の幹細胞が多いからです。どんな幹細胞が肺に多くあるのでしょうか?
肺に癌関連遺伝子が最も多いのは何故でしょうか?多種類の幹細胞が多いからです。成人呼吸器の上皮組織は表面積が70 m2と広大であり,多様な上皮細胞種と固有の特異的な組織構造を示します。複雑な呼吸器上皮組織を支える組織幹細胞もまた多様で複雑であります。気管,気管支では基底細胞とクラブ細胞が組織幹細胞で,気道クリアランスを支える細胞種を供給し,また互いに可塑性を持っています。クラブ細胞(club cell)は、クララ細胞(Clara cell)とも呼ばれ、終末細気管支と呼吸細気管支の移行部に存在する無線毛細胞ですが、細胞表面に短い微絨毛を有する。細胞質には分泌顆粒が存在し、開口分泌により放出される。クララ細胞は繊毛単純上皮に分布し、細気管支保護の為にしばしばムコ多糖であるグリコサミノグリカンを分泌する。代表的なムコ多糖はヒアルロン酸です。細気管支細胞は杯細胞の減少に伴い増加する。杯細胞とは(さかずきさいぼう)とは、眼、鼻、気道、消化管などの表面に存在し、粘液を分泌する細胞です。粘液が蓄えられている様子が杯のように見えることからその名があります。杯細胞の主な役割は、①粘膜表面を覆う粘液層を維持する②腸管の内面表面を潤滑にする③消化酵素などから保護する。杯細胞は、管を形成する器官の上皮細胞のなかに混じって単独に存在します。小腸にもかなりみられますが、とくに大腸に多く、気管の上皮にもみられます。クララ細胞の主要な機能の一つは多様な分泌物を分泌して細気管支上皮細胞を保護します。また、滑面小胞体にあるシトクロムP450酵素を用いて、肺に侵入した有害物質の無毒化も行う。クララ細胞は幹細胞としても振る舞い、増殖後に線毛細胞へと分化して細気管支上皮を再生する。炎症を抑える働きがある分泌物を出す。特別な物質を出して、気管支がくっついたり、気道がつぶれたりするのを防ぐ。 傷害を受けると,特に基底細胞が迅速に増殖期に入り,自己複製によって傷害部位を覆う.肺胞ではAT2 細胞(Ⅱ型肺胞上皮細胞)が組織幹細胞として知られており,自己複製を行うとともにAT1(Ⅰ型肺胞上皮)細胞を供給している.AT2 細胞(Ⅱ型肺胞上皮細胞)には亜集団が含まれ,特にWnt シグナルを活性化しているAT2 細胞が損傷再生によく貢献できる.最近,AT2 細胞がヘルペス感染によって肺線維症の原因になり間質性肺炎を起こします。特発性肺線維症(IPF)もAT2 細胞(Ⅱ型肺胞上皮細胞)がヘルペス感染によるものです。
間質性肺炎とKL6との関りは間質性肺炎において、KL-6が上昇することは、有名です。KL6とは?KL-6は、5000kDa以上のMUCムチンに属する糖蛋白のひとつで、肺では主にII型肺胞上皮細胞から産生され、間質性肺疾患における血清KL-6の上昇は、II型肺胞上皮細胞の障害や産生を反映する。ムチンとは動物の上皮細胞などから分泌される粘液の主成分である粘性物質であり、「粘素」と訳されることもある。ムチン はmucusを語源とする。Mucusは「粘液」という意味です。 実際には、分子量100万~1000万の糖を多量に含む糖タンパク質の混合物であり、細胞の保護や潤滑物質としての役割を担っている。KL-6は特発性肺線維症、過敏性肺炎、サルコイドーシス、膠原病関連間質性肺炎などの疾患活動性のバイオマーカーとしての有用なのはこれらの病気の原因はすべてⅡ型肺胞上皮細胞に感染したヘルペスウイルスの溶解感染で上皮細胞が崩壊してKL-6が血中に漏出したからです。KL-6は、主に肺胞Ⅱ型上皮細胞に発現するシアル化糖蛋白です。KL-6の臨床的意義はKL-6は、分子量100 万以上と巨大な分子量をもつ、シアル化糖蛋白です。血清KL-6 値は、特発性間質性肺炎症例の約 80%で高値を示す。KL-6は、特発性間質性肺炎の急性悪化時、活動性の評価、治療効果の判定に際して、極めて有用なマーカです。①通常、肺のⅡ型肺胞上皮細胞、呼吸細気管支上皮細胞などや、膵管、乳管などの腺細胞で産生されています。②間質性肺炎では炎症に伴って、Ⅱ型肺胞上皮細胞の障害や再生により、KL-6が過剰産生され血中で高値となります。③健常者やその他の肺疾患では高値を示さないため、間質性肺炎の診断マーカー、活動性の指標として使用されています。異常値を示す主な疾患や状態は①異常高値は特発性間質性肺炎、膠原病に関連した間質性肺炎、過敏性肺炎、放射線肺臓炎②検査値に影響を及ぼす要因は膵癌、乳癌、肺腺癌などでは高値を示す場合がある。
KL-6とSPA, SPDの間質性肺炎の血清マーカーとして比較は(1)感度と特異度については
KL-6以外にSP-A、SP-D、MCP-1が知られているが、KL-6が、間質性肺炎において最も感度(94%)、特異度(96%)が高いのです。(2)上昇のタイミングについては通常、間質性肺炎急性増悪の初期にSP-Dが上昇し、少し遅れてKL-6は上昇してくることが多い。またステロイド奏功時には、SP-D、KL6の順に低下する。血清マーカー上昇のタイミングがずれる機序としては、分子サイズの違いや、生理的環境下での存在形態の違いなどが挙げられている。(3)胸部CT所見との関係についてはKL-6は、すりガラス陰影の肺野面積に占める割合(5つの断面で測定)と相関するが、線維化病巣(牽引性気管支拡張の存在する区域の数)との相関がより強い。SP-A、SP-Dはすりガラス陰影として認められる胞隔炎の程度と相関があり、線維化病巣である蜂巣肺との広がりとの相関は低い。胞隔炎とは間質から始まった炎症が肺の実質に及び、胞隔炎(ほうかくえん)といって肺の中まで壊れていくため、最終的にその部分が膠原線維などで置き換わり、構造改悪を来した状態です。肺の組織を構成する「肺胞(はいほう)」というひとつの単位があります。これはブドウの房のようなイメージで、そのブドウの房のようなものひとつが小葉(しょうよう)などの呼び名で表されます。肺胞は直径約300μm(1μmは0.001㎜)で、約3億個存在し、肺胞を広げた総表面積は約70㎡に達します。その小葉などブドウの房同士の間を「間質」といいます。間質性肺炎ではその部分が病変の主体となり、炎症と線維化が起こります。線維化とは、「間質」に膠原線維と呼ばれる繊維ができてくることで、そのために肺胞が膨らみにくくなり硬くなってしまいます。それが肺の実質まで進展してきて肺実質そのものをつぶし、肺組織の構築が変わってしまい息ができにくくなるのです。
(4)KL-6と予後については特発性肺線維症27例の検討において、KL-6 1000U/mL以上の場合には、1000U/mL未満と比べて予後が不良であった。生存期間中央値は、KL-6値が高い群(1000U/mL以上)では18ヶ月、KL-6値が低い群(1000U/mL未満)では36ヶ月であった。この症例は間違ったステロイドホルモンを使ったからです。間質性肺炎の原因はヘルペスが肺胞Ⅱ型上皮細胞に感染して細胞が崩壊してシアル化糖蛋白であるKL-6が漏出しただけですから抗ヘルペス剤と漢方煎じ薬を用いれば確実によくなります。何故ならば肺胞Ⅱ型上皮細胞には幹細胞がありますからヘルペスで崩壊した肺胞Ⅱ型上皮細胞を新たに作り出すことができるからです。ARDSにおいて、生存例と比較して死亡例では、全経過を通じてKL-6値は高値を呈していた。ARDSは英語でAcute Respiratory Distress Syndromeで「急性呼吸窮迫症候群」と訳します。ARDSとは、重症肺炎や敗血症、外傷などの様々な疾患が原因で肺胞Ⅱ型上皮細胞が急速に崩壊してしまい重度の呼吸不全となる症状の総称です。肺に液体が貯留し、血液中の酸素レベルが異常に低下することで、息切れや呼吸の速さ、浅さ、皮膚の斑点やチアノーゼなどの症状を引き起こします。また、心臓や脳などの他の臓器が機能不全に陥ることもあります。ARDSは、慢性の肺疾患がある方だけでなく、肺にまったく異常がなかった方でも重篤な肺感染症で肺胞Ⅱ型上皮細胞が急速に崩壊してしまい高値なKL-6値が見られます。
(5)間質性肺炎以外にKL-6が上昇する疾患については肺胞蛋白症、ニューモシスチス肺炎、瀰漫性汎細気管支炎などでもKL-6は上昇する。肺胞蛋白症とは自己免疫性疾患といわれていますのでヘルペスが原因です。この世に自己免疫疾患が存在すればすべての生命は生まれることは無かったことでしょう。肺胞蛋白症が自己免疫疾患ではないのは肺胞に蛋白様物質が貯留する病気の原因はヘルペスが増えるために感染細胞が産生した栄養素やアミノ酸や塩基やタンパク質や糖分のすべてを利用し尽くされてしまうと生きられなくなった細胞が崩壊してしまい蛋白の崩壊産物であるアミロイドが不溶性ですから不溶性のアミロイドタンパクが肺胞に蓄積した病気が肺胞蛋白症であるからです。
ニューモシスチス肺炎とは、酵母様真菌であるニューモシスチス・イロベチイ によって引き起こされる肺炎である。真菌類は、キノコ・カビ、単細胞性の酵母、鞭毛を持った遊走子などの多様な形態を示す真核生物であり、菌界に分類される生物群である。遊走子とは無性生殖を行なう胞子の一種。鞭毛(べんもう)をもち水中で遊泳運動をする。藻類や下等な菌類に見られる。ニューモシスチス肺炎は正常な免疫能力を持つ場合発症することは希であり、化学療法やステロイド剤長期内服、後天性免疫不全症候群などによる免疫低下時に発症する、日和見感染症の一つである。瀰漫性汎細気管支炎とは、副鼻腔気管支症候群のひとつで、細気管支のヘルペス性の炎症により、慢性の咳嗽、痰、息切れを呈し、慢性副鼻腔炎を合併する呼吸器疾患。「瀰漫性」とは病変が一カ所だけにとどまらず、広範囲にわたっているということです。
KL−6が高値になるのは肺腺癌、乳癌、膵臓癌などの腺癌や、肺扁平上皮癌でも見られる場合があるので、癌を合併した間質性肺炎のKL−6が上がるのはすべての癌の原因もヘルペスであるからです。つまりヘルペスが原因で間質性肺炎と肺癌が合併することがあるのも当然であるのです。
健常者、有疾患対照群、間質性肺炎群(特発性間質性肺炎、過敏性肺炎、膠原病関連間質性肺炎)の合計547例を対象とした検討では、KL-6陽性率(カットオフ値は500U/mL)は、健常者0.5%、膠原病1.6%、肺炎3.1%、肺気腫2.4%、気管支拡張症10.7%および肺結核23.7%に対して、特発性間質性肺炎 95.0%、過敏性肺炎 89.7%、膠原病関連間質性肺炎では58.5%であった。カットオフ値とは、病態を識別するための検査や測定において、基準範囲を基本として正常とみなす範囲を区切る値です。特定の疾患に罹患している、または罹患するリスクがあるかどうかを分ける値として用いられます。
KL-6と肺癌の関りはKL-6は肺疾患で血中に流出する抗原で、肺腺癌や乳癌、膵癌などの悪性腫瘍でも高値となることがあり、肺癌と関連があります。KL‐6とは広島大学の河野修興先生が肺癌の関連抗原を探していたときに見つけた間質性肺炎の患者さんに高く検出されるバイオマーカーです。特発性肺線維症(IPF)などの間質性肺疾患(ILD)だけでなく、亜急性過敏性肺炎でもかなり高くなることが知られています。
KL-6は、主に肺胞Ⅱ型上皮細胞に発現するシアル化糖蛋白で、特に薬剤性間質性肺炎や特発性間質性肺炎で高い陽性率を示します。間質性肺炎では急性増悪時に著明に上昇するため、間質性肺炎の活動性の評価に有用です。
一方、肺腺癌や乳癌、膵癌などの悪性腫瘍でも高値となることがあり、進行すると50%前後の症例が異常値を示すことに注意が必要です。また、肺胞蛋白症、ニューモシスチス肺炎、瀰漫性汎細気管支炎などでも上昇します。
KL-6が異常値を示したからといって間質性肺炎と決めつけることはできません。
肺の組織の成り立ちについて肺の組織を構成する「肺胞(はいほう)」というひとつの単位があります。これはブドウの房のようなイメージで、そのブドウの房のようなものひとつが小葉(しょうよう)などの呼び名で表されることもあります。その小葉などブドウの房同士の間を「間質」といいます。間質性肺炎ではその部分が病変の主体となり、炎症と線維化が起こります。線維化とは、膠原線維と呼ばれる繊維ができてくることで、そのために肺胞が膨らみにくくなり硬くなってしまいます。それが肺の実質まで進展してきて肺実質そのものをつぶし、肺組織の構築が変わってしまいます。

つまり、間質から始まった炎症が肺の実質に及び、胞隔炎(ほうかくえん)といって肺の中まで壊れていくため、最終的にその部分が膠原線維などで置き換わり、構造改築を来した状態です。その壊れた部分が修復されていくときにも、余計に膠原線維が出てきて過剰な修復が進行するため、間質性肺炎という病態ができるのだろうと考えられています。肺の組織を構成する「肺胞(はいほう)」というひとつの単位があります。これはブドウの房のようなイメージで、そのブドウの房のようなものひとつが小葉(しょうよう)などの呼び名で表されます。
その小葉などブドウの房同士の間を「間質(かんしつ)」といいます。間質性肺炎ではその部分が病変の主体となり、炎症と線維化が起こります。線維化とは、膠原線維と呼ばれるものができてくることで、そのために肺胞が膨らみにくくなり硬くなってしまいます。それが肺の実質まで進展してきて肺実質そのものをつぶし、肺組織の構築が変わってしまいます。

つまり、肺胞細胞に感染したヘルペスが細胞を壊死させて間質に癒合することから始まった炎症が肺の実質に及び、胞隔炎(ほうかくえん)といって肺の中まで壊れていくため、最終的にその部分が膠原線維などで置き換わり、構造改築を来した状態です。その壊れた部分が修復されてもヘルペスは殺すことができないので次の肺胞細胞に感染していくときにも、同じように余計に膠原線維が出てきてさらに両肺で6億個もある肺胞細胞にヘルペスが感染していくと呼吸が出来なくて死ぬことがあります。間質肺炎はこの世には存在しない自己免疫疾患と言われる難病とされています。実は自己免疫疾患はすべてヘルペスが原因の病気ですから肺胞細胞の崩壊の度合いが少なければ抗ヘルペス剤と免疫を上げる「漢方煎剤」で確実によくなっていきます。
癌血管転移のメカニズム
がん細胞はどんどん増え続けるだけでなく、周囲の組織へ広がり(浸潤)、血液やリンパの流れに沿って、遠くの臓器へ引っ越して、やがてまたそこで「すみか」を作って大きくなります。これが転移と呼ばれる現象です。転移があると、がんができた場所(原発巣)のコントロール(外科治療、化学療法、放射線治療など)がうまくいっても、最終的に転移巣(がん細胞が転移をした場所)が大きくなって、治療が不成功に終わります。
まさに、『転移を制すものはがんを制する』というくらい、転移は悪性化の最たるものですここでは、最も頻度の高い血管を介した転移の仕組みについて、簡単に解説します。
原発巣で大きくなったがんの固まりは一様ではなく、その中には高い運動能力や周りの組織を破壊する能力(細胞の移動を妨げるがんの周りのコラーゲンなどの繊維組織を分解する酵素を作り分泌する能力など)をもった一群の細胞がいます。これらの細胞が周囲へ浸潤して血管壁に近づき、血管内に侵入します。外側から血管の内側に侵入した細胞群(2-10個)は血管内で血液細胞と塊を作り、血流に乗って、肺、肝臓、脳や骨髄などの転移先の臓器へ運ばれます。この血液内のがん細胞はがんの初期から存在し、患者さんの血液1ml中に5~1,000個流れているようです。やがて、転移先の血管の内側に接着した細胞塊は大きくなり、血管を内側から破って臓器内に侵入し、新たな「すみか」である転移巣を作っていきます。「すみか」を作るには酸素と栄養が必要で、直径3mm以上の大きさになるためには新たな血管を作って補給路の整備をします。こうして転移先で大きくなったがん細胞の塊は種々の画像検査で見えるようになり、臨床的に転移と診断されます。
興味深いことに、原発巣と転移先には“種と土”のような相性があり、大腸がんは肝臓へ、肺がんは脳へ、乳がんや前立腺がんは骨(骨髄)へよく転移します。その理由の一つとして、転移先の組織が分泌するケモカインとよばれる物質と、がん細胞の表面に発現するケモカインと結合する受容体が働き合って、特定のがん細胞が特定の組織に行きやすくなるのだと考えられています。
癌死はどのようにして起こるのでしょうか?癌死を決定するのも増えたヘルペスの数によって左右されます。ヘルペスの数が増えれば増えるほど一個の細胞に癌を起こす2種類の合計800個の癌関連遺伝子の癌化の数が増えていき癌の悪性度と進行度が高まり上の表にあげたように致死性の癌になっていくのです。

致死性の癌になっていく順位を決定する最も大きい因子は何でしょうか?
がんを最初に生み出すのはヘルペスが癌関連遺伝子の癌原遺伝子の一個と癌抑制遺伝子の一個を癌化すれば一個の癌細胞が生まれます。この癌細胞が分裂するときに絡まって一対の23本の染色体はほぐれてバラバラの1本になった46本の染色体、さらに倍加して接着していた姉妹染色分体が分離して1本のバラバラになった92本の染色体になるのでヘルペスはこのバラバラになった一本鎖の染色体の状態の時に簡単に自分のゲノムを組み込ませ部位特異的組み換えによってヘルペスウイルスは細胞のDNAに入り込むときに使う機構を用いて自分のDNAのゲノムを細胞のゲノムに入れ込み細胞の塩基配列を簡単に組み換えて遺伝子の突然変異を起こしてしまいます。たまたまこの突然変異を起こした遺伝子が癌原遺伝子の一個と癌抑制遺伝子の一個であれば癌細胞が新たに一個生まれることになります。さらに細胞が分裂するためには細胞には細胞周期という4つのG1期、S期、G2期、M期があります。細胞周期が1周り分はG1期→S期→G2期→M期です。増殖中の細胞の場合分裂から分裂までの間が細胞周期1回分です。Gはgap(間隙)の略語です。G1期→S期→G2期をまとめて間期と言います。M期は融資分裂と細胞質分裂が行われます。ヘルペスはなぜ生きた細胞に感染せざるを得ないのでしょうか?ヘルペスは増殖分裂するためのエネルギーはもとより、エンべロープ、スパイク、テグメント、カプシドなどを作るための原料のみならずさらに自分の2本鎖直鎖DNAを複製する材料をすべて一切持っていないのです。持っているのはDNAに設計されている遺伝子情報だけです。エンべロープ、スパイク、テグメント、カプシドなどを作る遺伝子情報を発現させる蛋白質、脂質、糖などの原料を細胞から盗み出して利用するために細胞に感染しているだけです。それどころかヘルペス自身の遺伝子を構成しているヌクレオチドの原料である4種類の塩基であるアデニン、グアニン、チミン、シトシンもDNAポリメラーゼ5単糖(リボース)もリン酸もDNAポリメラーゼを作るアミノ酸もすべて感染した細胞から盗み取るために細胞に感染しているだけなのです。盗み取られた細胞はどんなに疲弊しきるか想像できますか?一個の細胞でヘルペスが1000個も増えてしまうと溶解感染で溶け死んでしまうのも当たり前のことなのです。しかもできるだけ多くの子孫(ビリオン)を増やすために細胞が死ぬまで利用しまくるのです。さらに増えたヘルペスはあらゆる細胞のゲノムに入り込んで遺伝子の塩基の並びを変えたり染色体の数の増減をもたらしてあらゆる遺伝子病をこっそり作りその果ては癌原遺伝子やがん抑制遺伝子を癌化させてしまい癌細胞が生まれるのです。
細胞が2個になるのに50時間かりますがヘルペスが2個になるのは感染細胞にもよりますが1分以内なのです。世界中で癌細胞の発がんからから増殖、転移、悪液質、癌死までのすべてを支配しているのはヘルペスであることを知っているのは私だけなどこの一流大学病院を受診しても原因不明だと言われ人生に絶望して3回も自殺未遂を試みた馬鹿な男でしたが自分の病気を知るために自分の病気を治すために医者になってから私の病気のすべてを解明できたのです。現代医療はヘルペスがすべての病気の原因であることを認めないどころか免疫を押さえて症状を減らして安心させその裏ではこっそり患者をだまして病気の原因であるherpesを増やして最後は癌まで増やして最近亡くなられた近藤誠先生ではないのですが患者を殺す医療に励んでいるだけです。残念です。
それは15歳からヘルペス性脳炎になってしまい79歳になった今でも苦しんでいるので死ぬまで病気のすべての原因は何かを追究し続けてヘルペス性脳炎どころか自己免疫疾患のみならず癌の原因までもヘルペスであることを理論的にも臨床的にも証明することができたのです。癌はヘルペスが原因であることは私には明々白々であることなのに何故誰も知らないのでしょうか?難病中の難病とされている病気が癌ですがもう一つの最後に残された難病は精神病であり精神病の原因も広い意味でherpes脳炎なのです。ヘルペスが脳に感染して生じた脳の神経変性疾患と言い換えてもいいのです。私自身が経験してきたので誰よりの良く知っているのです。何故ものすごい数のヘルペスは人のすべての40兆個もある細胞に感染して癌を含めてあらゆる難病を作り続けるのかを説明していきましょう。
ヘルペスの増殖スピードに興味を持っている癌の研究者は誰もいません。従って増えたビリオンが10億、100億、1000億とherpesが増え続けるときに癌細胞自身がその結果、人も餓死するのも当然だといとも簡単に理解できるでしょう。又生命にかかわる細胞に癌が転移してその結果、人も癌死するのは当たり前のことであることが理解できます。
胃癌が肺に転移した時は転移した胃癌の細胞は胃細胞癌なのか肺細胞癌のどちらの細胞の癌になっているのでしょうか?答えは胚細胞癌です。逆に肺癌が胃に転移した時には転移した肺癌細胞は時間がたつにつれて胃癌に変わっていくのです。その理由は自分で考えてください。詳しい答えは後述します。
細胞周期とはなんでしょうか?細胞分裂周期とも言います。細胞分裂とは親細胞が二つの娘細胞に増殖し分裂する過程です。細胞周期(cell cycle)は、一つの細胞が二つの娘細胞を生み出す過程で起こる一連の事象とその周期のことをいう。細胞周期の代表的な事象として、ゲノムDNAの複製と分配、それに引き続く細胞質分裂がある。細胞周期は、光学顕微鏡での観察に基づき、間期(interphase)とM期(M phase)との二つに大きく分けられる。間期はさらにG1期、S期(synthesis、DNAの合成期)、G2期の3つに分けられる。M期は有糸分裂と細胞質分裂によって構成される。有糸分裂では遺伝子が全く同じ姉妹染色分体が細胞の両極に分かれ、引き続く細胞質分裂では細胞質が割れて2つの細胞が生み出され細胞分裂が終わります。因みに一時的にもしくは可逆的に分裂を停止した細胞は、G0期(静止期⦆と呼ばれる静止期に入ったとされる。G0期(静止期⦆とは何でしょうか?
増殖能力は保ちつつも細胞分裂を停止している状態のことです。G0期の別名は?G0期の別名は休止期,休眠期,静止期,quiescent,dormantとも表現される.細胞周期(G1/S/G2/M期)以外にある状態です。 実は、体内に存在する細胞のほとんどはG0期にとどまっており、増殖を停止しています。 これは細胞が接触しているために生じる性質として考えられておりコンタクトインヒビション(細胞の接触障害)と呼ばれています。動物の冬眠(hibernation)に類似し,代謝が低下したいわば省エネモードともいえるでしょう。
間期(interphace)とは何でしょうか?細胞が分裂し、生じた娘細胞が再び有糸分裂を開始するまでの間、つまりM期と次のM期の間を間期(interphase)と呼ぶ。細胞の成長、物質の吸収、生合成、遺伝情報と全ての細胞小器官の複製、また代謝など、細胞としての機能はこの時期に行われる時に同時にへルペスも増殖分裂を激しい勢いで来り返しているのです。真核細胞の多くは大半の時間を間期に費やし、次の細胞分裂(M期)に備える。勿論ヘルペスの増殖分裂には間期も細胞分裂(M期)もありません。間期では、クロマチンは核膜に囲まれた細胞核の中に分散しており、個々の染色体を識別することはできない。核小体は核内構造のひとつとして確認できる。紡錘糸はまだ観察されないが、中心体は核周辺に観察される。核小体とは真核生物の細胞核の中に存在する、分子密度の高い領域で、rRNAの転写やリボソームの構築が行われる場所のことで、一般に光学顕微鏡で観察できる。 直径1〜3μm程度。 仁、核仁とも言われる。
放射性同位体(RI, radio isotope)を用いて同調細胞のDNA合成を経時的に追跡することで、間期は、G1期、S期、G2期の3段階に分けられることが明らかになった。各期は細胞周期チェックポイントで完了が確認されてから次の期間へと進行する。各期間と間期全体にかかる時間は細胞の種類や生物の種類によって様々である。一般に哺乳類の成体の細胞で、間期は20時間ほどであり、細胞分裂全体のほぼ90%の時間を占めるM期(細胞分裂期)は25時間かかるので一回の細胞周期にかかる時間は45時間くらいです。
G1期とはM期が終わり、新しくDNA合成が始まるまでの期間は、間期における最初の期間であり、G1期(Gはgapを意味する)と呼ばれる。G1期は別名(Gをgrowthと読んで)成長期とも言われるのはDNA合成が始まるのは細胞の生長期の始まりでもあるからです。この期間中、M期では顕著に低かった細胞の生合成活性が再び高まりherpes増殖のための生合成活性が高まりにもなります。G1期では、次のS期で必要とされる種々の主に細胞のDNA複製に用いられるかつherpesのDNA複製に用いられる酵素が合成されます。また細胞小器官の合成も盛んで、関連する構造タンパクと酵素が多量に細胞のためにもヘルペスウイルスのためにも消費されるため細胞内の代謝が活発な期間でもある。G1期はさらに4つの小期に分けられる。①コンピテンス(g1a)②エントリー(g1b)③プログレッション(g1c)④アセンブリ(g1d)がG1期の4つの小期になります。
これらの小期は成長因子、栄養供給、温度、その他の阻害因子であるherpesウイルスに大きく影響を受けうる。S期に入る前にG1期を中断し休眠状態のG0期に入る細胞もある。G1期の長さは様々で、同種の生物でも細胞によって異なるが、消化器官のような24時間毎に分裂を繰り返しているような活発なヒトの細胞では、G1期に約9時間かかる。G1期の終わりには細胞周期チェックポイントがある。これはDNAに欠陥がなく、細胞の機能が正常なことを確認する一連の安全機構である。機能的にはサイクリン依存性キナーゼ(Cyclin Dependent Kinase; CDK)がこの役目を果たしている。G1期CDKタンパクは様々な遺伝子に対して転写因子を活性化する。これらの遺伝子の中にはDNA合成タンパク質やS期CDKタンパク質に対応するものも含まれている。細胞周期チェックポイントとは、細胞が正しく細胞周期を進行させているかどうかを監視(チェック)し、異常や不具合がある場合には細胞周期進行を停止(もしくは減速)させる制御機構のことである。細胞自体がこの制御機構を備えている。一回の細胞分裂の周期の中に、複数のチェックポイントが存在することが知られており、これまでに①G1/Sチェックポイント、②S期チェックポイント、③G2/Mチェックポイント、④M期チェックポイントの4つがある。このチェックポイントの機構は正確な遺伝情報を娘細胞、ひいては子孫に伝達するための、生命にとって根源的な役割を果たしており、このチェックポイントの機構の異常はヒトなどのがん発生の主要な原因のひとつです。
S期とはG1期に続くS期(Synthesis phaseの略)は、染色体DNAが複製される時期である。S期では、DNAヘリカーゼが2重鎖DNAを開裂して1本鎖を作り、続いてDNAポリメラーゼが相補的塩基対を結合させることで2本の2重鎖DNAが生成される。
DNAヘリカーゼとは何でしょうか?DNAヘリカーゼ(helicase; ヘリケース)は核酸のリン酸エステル骨格に沿って動きながら絡み合う核酸をほどく酵素の総称である。DNAの2本鎖をほどくものを特にDNAヘリカーゼ(DNA helicase)、RNAの二次構造をほどくものをRNAヘリカーゼ(RNA helicase)と呼びます。DNAヘリカーゼの機能はDNA複製、DNA修復、DNA組換え、転写、翻訳、スプライシングなど、遺伝情報を扱う様々な過程で対合している核酸をほどく必要がある。そこでヘリカーゼは、ATPやGTPを加水分解して得られるエネルギーを使って塩基間の水素結合を解消し、DNAの二重らせんや二次構造を取ったRNAなどをほどく働きをしている。ヘリカーゼは、片方の鎖に沿って種類毎に決まった方向に動きながら働く。DNA合成が完了し、全ての染色体が複製されたところでS期は終了する。S期の間に細胞内のDNA量は実質2倍になる。S期ではRNA転写とタンパク質合成の速度は非常に低い。しかし、ヒストンは例外的で、ほとんどのヒストンがS期に作られる。中心体もS期に複製される。DNAの複製と中心体の複製は独立に行われるが、その進行には多くの共通の因子が関係している。結果的に細胞分裂に必要な細胞内の遺伝物質の複製はS期で完了する。中心体とは中心体は染色体を引っ張り分離します。染色体が引っ張られた先では、また新しい細胞が作られ、分離された染色体が取り込まれていきます。 このように、中心体は細胞分裂において、紡錘糸によって染色体を分離します。S期ではDNAの損傷が頻繁に起こるが、複製の完了と共にDNA修復が始まる。修復が不完全な場合はS期細胞周期チェックポイント機構で検知され、細胞周期が停止される。この段階を通過するとほとんどの細胞は細胞周期を途中で停止することはないのです。
G2期とはDNA複製が完了してから、M期に入るまでの期間をG2期と呼ぶ。G2期では再び盛んなタンパク質合成が行われ、主に有糸分裂に必要な微小管が作られる。一般に間期の中ではG2期が最も短く、例えばヒトの細胞では多くの場合4~5時間で終了する。G2期にはG2/Mチェックポイントがあり、細胞がM期(有糸細胞分裂期)に進めるかどうか判断している。
M期とはM期(Mitotic phaseの略で訳は有糸細胞分裂期)には、有糸細胞分裂(mitosis)と細胞質分裂(cytokinesis)が行われる。有糸分裂(mitosis)は、ほとんどの細胞において複雑な分裂工程ですが約1時間程度で終了する。有糸分裂は、染色体の動態を光学顕微鏡で観察すると、前期・前中期・中期・後期・終期の5期に分けられる。前期では、①染色体の凝縮(染色体凝縮)が起こり、この時期に②染色体が顕微鏡下で観察されるようになる。中期にはいると、③核膜が消失し、④染色体が赤道面上に並ぶ。⑤紡錘体もこの時期に完成する。後期では、⑥セントロメア付近で結合していた姉妹染色分体が、紡錘体に引っ張られるような形で、分離し、極方向に移動を開始する。終期では分離を終えた⑦染色分体が脱凝縮し、⑧その周囲に核膜が再形成される。また、この時期から⑨細胞質分裂が始まり、➉細胞分裂が終了する。
静止期(G0期)とはG0期は、細胞分裂も分裂の準備も行われていないG1期が延長している状態であるとも、細胞周期から分かれた活動停止状態とも捉えられている。また、神経細胞や心筋細胞などは、細胞分化の果てに有糸分裂の後分裂を止め、成熟し、残りの寿命期間を本来の機能を一切分裂することなしに発揮し続ける。これらの細胞にとってG0期は細胞周期外の非分裂状態にあることからG0期は「有糸分裂後」とも言われることもある。細胞質分裂をしない多核筋細胞もG0期にあると表現される。「有糸分裂後」という用語は、時折G0期と細胞の老化の両方を示す際に使われることがある。多細胞真核生物における非増殖性細胞は一般的にG1期からG0期に入り、長期に、時には神経細胞の場合などは特に無制限にG0期にとどまることがある。完全に細胞分化した細胞のほとんどはG0期に入る。細胞の老化は、子孫細胞が成長できなくなるようなDNAの損傷や劣化に反応してなる状態である。細胞の老化とは、損傷を受けた細胞を自己破壊するアポトーシスの生化学的代替手段ともいえる。
細胞の老化とは何でしょうか?細胞が分裂を停止し、増殖できなくなった状態が不可逆的に引き起こされることです。ゲノムの不安定化などによって引き起こされ、細胞ががん化することを抑制する防御反応であると考えられている。ゲノムの不安定化とはゲノム(DNA)は放射線や化学物質などさまざま要因によって日々傷ついているが、「DNA修復機能」によって修復されている。 ところが、この修復機能に異常があると、正常に修復されず、変異が起きやすい状態をゲノム不安定性となるのです。ヒトの初代培養細胞に「ヘイフリック限界」と呼ばれる分裂回数の制限があることが発見され、細胞老化は狭義にはこの限界に達した細胞の状態を指した。生体内 (in vivo) の細胞でも、自己防御のための積極的な細胞老化が起こりこの細胞老化は未成熟細胞老化と名付けられたが、人工的な条件下 (in vitro) で起こるヘイフリック限界よりも、生物学的な意義が認められ、「細胞老化」が未成熟細胞老化を指す場合もある。未成熟細胞老化はさまざまな生物学的ストレスにより引き起こされる。例えばテロメアが短縮すると染色体が不安定になり、がん化の原因となる。
染色体が不安定とは癌や先天性疾患において染色体の数の異常、欠失、転座などが広範囲に見られる染色体の構造異常のこと。正常な細胞は細胞分裂の際に染色体を倍加させ、細胞質の分裂に伴い染色体を均等に分配することができ、結果的に1つの細胞に46本の染色体が備わるようになる。テロメアが短縮すると染色体が不安定になり、がん化の原因となるのでテロメアの長さを監視する機構があり、一定以上短くなると一時的な細胞老化が誘導される。またヘルペス感染によってDNAの切断が生じた場合も、細胞周期を停止させ細胞分裂が起こらないようにし、その間に染色体の修復を行う。それでも復旧できなかった場合は不可逆的な細胞老化状態に入るか、アポトーシスによって排除されるが、これらの機構を逃れた細胞はがん化する。このように細胞老化の多くの原因はDNA損傷によって誘導される。DNA損傷は放射線や変異原、酸化ストレスによって引き起こされる。DNA損傷が即遺伝子を突然変異して癌になることはないのです。確かに遺伝子はDNAから構成されていますがDNA損傷と発癌は別問題です。これを癌研究者は同一視していますが間違いです。
細胞周期の制御(細胞周期の進行)は細胞周期エンジン(Cdk/サイクリン複合体)によって制御されている。細胞周期エンジンとはCdk(cyclin-dependent kinase, サイクリン依存性キナーゼ)とサイクリン(cyclin)の複合体です。Cdk/サイクリン複合体は、細胞周期を前に進めることから、自動車のエンジンに倣って細胞周期エンジン(cell cycle engine)と呼ばれます。
サイクリン(cyclin)とは何でしょうか?細胞がいつ分裂しいつ分裂すべきでないのかを制御します。サイクリン (Cyclin) は、真核生物の細胞において細胞周期を移行させるためのエンジンとして働く蛋白質のひとつ。現在までに哺乳類では20種類以上のサイクリンが見つかっている。サイクリンと細胞周期の関わりは細胞はG1→S→G2→M期からなる細胞周期を回転させることにより増殖をする。細胞周期の回転において自動車のエンジンと同じ役割を果たすのがサイクリンとサイクリン依存性キナーゼ (Cyclin Dependent Kinase; CDK) と呼ばれる蛋白質との複合体であり、これらは複合体を形成してのみ働く。サイクリンはCDK(サイクリン依存性キナーゼ)の活性発現に必要であり、サイクリンは調節サブユニットと呼ばれる。細胞内には複数の種類のサイクリン及びCDK(サイクリン依存性キナーゼ)が存在し、細胞周期の回転にはサイクリンA, サイクリンB, サイクリンD, サイクリンEも関与している。その他のサイクリンは転写制御などの役割を果たしています。CDKは34–40 kDaの比較的小さなタンパク質で、ほぼキナーゼドメインのみから構成される。CDKはサイクリンと呼ばれる調節タンパク質に結合するのは、サイクリンがなければCDKはほとんどキナーゼ活性を持たず、サイクリン/CDK複合体を形成することで活性型キナーゼとなる。
各細胞周期の回転において細胞はサイクリン及びCDKの組み合わせを変えて使い分けている。例えばサイクリンEとCDK2の複合体はG1/S期に働くが、G1期になるとサイクリンEの発現量が増加して細胞周期の進行に関与するが、S期になるとユビキチン-プロテアソーム系により分解されてしまう。その後の細胞周期の進行は他のサイクリン-CDK複合体が担うので問題はない。また、細胞周期に依存して発現量が変化するのはサイクリンの方だけで、CDKの発現量は変化しない。
Cdk (CDK)とは何でしょうか?サイクリン依存性キナーゼ(cyclin-dependent kinase、CDK)である。真核生物の細胞周期のエンジンとして働くプロテインキナーゼである。 増殖細胞ではDNA損傷やDNA複製,染色体分配などの異常の結果によりゲノムが維持できなくなると,エンジンであるCdk(CDK)は活性化されず細胞周期の進行が停止し細胞死が誘導のです。動物細胞では、複数のCdk/サイクリン複合体が細胞周期の進行に関わっている。G1期からS期へ進むにはCdk2/サイクリンE複合体、S期からG2期への進行にはCdk2/サイクリンA複合体、G2期からM期への移行にはCdk1/サイクリンB複合体の活性が必要である。因みにCdk1はCdc2とも呼ばれます。必要なときに必要な複合体のみ活性化するために、細胞内では、各サイクリンの転写の制御やユビキチン依存的な分解、Cdkはリン酸化・脱リン酸化などの修飾による活性の制御が行われているのです。
細胞周期チェックポイントは細胞周期の進行を正常に行えるか監視するポイントがあり、これを細胞周期チェックポイント機構という。細胞周期チェックポイントとは、細胞が正しく細胞周期を進行させているかどうかを監視(チェック)し、異常や不具合がある場合には細胞周期進行を停止(もしくは減速)させる制御機構のことである。細胞自体がこの制御機構を備えている。一回の細胞分裂の周期の中に、複数のチェックポイントが存在しており、これまでにG1/Sチェックポイント、S期チェックポイント、G2/Mチェックポイント、M期チェックポイントの4つがよく解析されている。この細胞周期チェックポイント機構は正確な遺伝情報を親細胞から娘細胞、ひいては子孫に伝達するための、生命にとって根源的な役割を果たしているのです。この機構の異常を引き起こすのはヘルペスウイルスでありヒトなどのがん発生の主要な原因のひとつなのです。これらのチェックポイントでは細胞周期エンジンであるCdk(cyclin-dependent kinase, サイクリン依存性キナーゼ)とサイクリン(cyclin)の複合体の働きを阻害することによって細胞分裂周期の進行を止めることができます。因みに人の体では一生の間に約一京回の細胞分裂が行われています。
何故細胞周期のチェックポイントは極めて厳格です。何故でしょうか?ほとんどすべての生体分子であるタンパク質やRNAなどは傷ついても処理してしまっても生合成で簡単に作れる。しかしDNAだけは簡単に合成できないので傷がDNAにつくとなんとか元のままのDNAに復元する必要があるのです。仮にDNAにも傷がつくことが有りDNA損傷には塩基損傷とDNA鎖切断の二つに区分できます。細胞に備わった生命を支配する傷ついたDNAを修復するシステムは、あらゆる種類のDNA損傷を事実上100%治癒する力がある。癌はDNAの損傷で起こるのと現代医療は言いふらしていますが間違いです。癌はヘルペスによって二種類の癌関連遺伝子が突然変異して一個の癌細胞が生まれで次々と8百個もある癌関連遺伝子の突然変異が積み重なって原発巣の癌細胞塊が大きくなり浸潤が播種になり遠隔転移もherpesが原因で癌が拡大していくのです。癌の血管新生も癌患者が悪液質になるのもherpesが無限大増殖分裂を繰り返すために細胞から栄養のすべてを奪い取るためにherpesによって奴隷にされた細胞のなせる結果なのです。癌転移についてはここを読んでください。
細胞は、その性状や生体内での役割に応じて、それぞれ決まった周期で細胞分裂を繰り返し増殖している。この、一回の分裂増殖の周期を細胞周期と呼び、例えばいくつかの種類のヒト培養細胞の細胞周期は約24時間である。しかし、細胞にX線を照射してDNAに損傷を起こすと、この周期が長くなるのです。何故でしょうか?それは細胞にはDNA損傷などの遺伝子異常が起きると、損傷した無駄なDNAを作る前にそれを検知して細胞周期を一旦停止させる機構が存在することが発見され、この遺伝子異常を監視し細胞周期を止める機構は細胞周期チェックポイントと名付けられ、略してチェックポイントともいわれます。DNAは特別な分子で細胞分裂周期においてのみでしか作れないので細胞分裂周期のチェックポイントは極めて厳しいのです。
細胞周期チェックポイントの役割は、①遺伝子(DNA)に損傷がないか(DNA損傷チェック)②DNA複製が正常に行われているか(DNA複製チェック)③有糸分裂中に、複製された染色体の分離が正しく行われるか(スピンドルチェック)などを監視しており、これらに異常が検知されると、チェックポイント制御因子と呼ばれる複数の分子群が活性化されて、細胞周期の進行を遅らせ、停止させる。スピンドルチェックとは何でしょうか?スピンドルとは紡錘という意味があります。スピンドルチェックポイントのターゲット(目標)のスピンドルは紡錘体と訳します。紡錘体という意味は糸を紡ぎ巻き取る小さく細長い円柱形の両端がとがった用具に似た形のものを紡錘体(ぼうすいたい英語でspindle)と言います。紡錘体は、真核生物の細胞分裂において、姉妹染色分体を娘細胞へ分離するために形成される細胞骨格構造です。姉妹染色分体とは姉妹染色分体(sister chromatids)とは、DNA複製後にできる、全く同じ遺伝情報をもつ2本の染色分体のことをいう。 複製後の染色体は一対の姉妹染色分体から構成されると言い換えることもできる。
紡錘体と微小管の関係は細胞質分裂に際して現れる赤道に染色体が整列して、さらに対になっていた姉妹染色分体が二つに分離する時に関わる微小管は紡錘体及び紡錘糸と呼ばれるのです。又染色体が両方向に線路に沿って移動する時の線路は細長い微小管であり実際は線路ではなく微小管に沿って移動しているといいます。
遺伝学的に同一な娘細胞を作り出す過程である有糸分裂の際に形成される紡錘体(微小管»は、mitotic spindle(有糸分裂紡錘体)と呼ばれる。また、母(親)細胞の染色体の半数を含む配偶子を形成する過程である減数分裂の際に形成される紡錘体は、meiotic spindle(減数分裂紡錘体)と呼ばれます。紡錘体は染色体に加えて、数百のタンパク質から構成されている。微小管はこの装置に最も豊富に含まれる構成要素です。細胞分裂期の微小管の働きは二極性の紡錘体を形成し、赤道面に並んだ染色体を細胞の両極に引き寄せるとともに、細胞表層に作用して分離した染色体の間に「分裂溝」と呼ばれるくびれを誘導することです。
S期に複製された染色体は全く遺伝子が同一である姉妹同士が対合して姉妹染色分体を形成します。この姉妹染色分体同志の対合は間期を通じて維持されます。有糸分裂初期から染色体有糸分裂期へ進行すると姉妹染色分体は凝縮を開始し、核の周辺では紡錘糸の形成極である中心体が二分割し、それぞれが核の両端へ移動をはじめます。核膜の崩壊に伴い紡錘糸と姉妹染色分体との活発な相互作用がおこります。姉妹染色分体の動原体にはそれぞれの極から伸長する紡錘糸が接続し、染色分体は紡錘糸に依存した運動により細胞の中心にある赤道面上に移動します。動原体とは動原体またはキネトコア( kinetochore)は、真核生物の細胞において、複製された姉妹染色分体に結合する円盤型のタンパク質構造であり、細胞分裂時に姉妹染色分体を引き離すために紡錘糸が結合する部位である。キネトコアはセントロメア上で組み立てられ、有糸分裂と減数分裂時に染色体を紡錘体の微小管ポリマーへ連結する。また、キネトコアのタンパク質は姉妹染色分体をつなぎとめておくのを助け、染色体の編集にも関与するすべて姉妹染色分体の対合が無くなり別れ適切に紡錘糸と接続し赤道面上に整列すると、有糸分裂後期に移行し姉妹染色分体は解離します。有糸分裂前中期には、ある染色分体は両極から伸長した紡錘糸との接続、さらに赤道面上へ移動を完了している一方で、他の染色分体は紡錘糸との接続が完了していない状況がおこり得ます。このような状況では赤道面上にある姉妹染色分体は解離しません。すべての姉妹染色分体が赤道面に整列するまで姉妹染色分体の解離を抑制する機構、すなわちスピンドルチェックポイントが機能しているからです。
スピンドルチェックポイントはどのようにして姉妹染色分体の解離を抑制するのでしょうか?姉妹染色分体の対合はコヒーシンとよばれるタンパク質複合体により維持されています。有糸分裂後期にいたり姉妹染色分体解離する際にはコヒーシンの分断が必要です。セパレースは特異性の高いタンパク質分解酵素で、コヒーシンのサブユニットを分断します。セパレースの活性はその阻害因子、セキュリンにより制御されます。細胞周期のほとんどの時間で、セキュリンはセパレースに結合し、その活性を抑制します。有糸分裂後期になるとセキュリンはユビキチン化依存的に分解され、これにともないセパレースが活性化されます。すなわち、セキュリンの分解のタイミングが、姉妹染色分体の解離の時期を決定します。

チェックポイント制御因子が活性化されると、その異常の原因が取り除かれるまで、細胞周期が停止した状態になります。この間に、例えば軽度のDNA損傷の場合には、DNA修復機構が働くことで損傷が修復されます。そして異常が完全に取り除かれたと検知された時点で、チェックポイントの働きが可逆的に解除され、再び細胞周期が進行します。このように細胞周期チェックポイントは、細胞分裂の過程で異常が生じた場合に、細胞周期を一旦停止させて異常の原因を取り除くことで、遺伝子異常が子孫に伝わらないようにする役割を果たしているのです。細胞周期チェックポイントは単に細胞周期の異常を監視しているだけではなくDNA損傷が見つかればDNA修復機構を働かせ細胞周期を一旦停止させて異常の原因を取り除くことで、遺伝子異常が子孫に伝わらないようにする役割を果たしているのです。
また一方で、重度のDNA損傷の場合などDNA修復機構でも完全な修復が出来ない場合、チェックポイント活性化に続いて、その細胞がアポトーシスを起こして死滅させる仕事も行っているのです。このアポトーシス機構は、遺伝子異常を起こした細胞が「自殺」することで、異常な細胞を後世に残さないようする役割を果たしているのです。細胞周期チェックポイントは、その細胞が損傷修復を経て再び増殖に向かうか、アポトーシスを起こすかというスイッチの制御にも関与しているのです。
チェックポイント機能に異常がおきると、内因性、外因性のDNA損傷によって、細胞は正確な親細胞のコピーである娘細胞を作れなくなる場合が多くなります。たとえば、チェックポイント機能の不良により生存に必須な遺伝子に損傷が起きた場合、その細胞は娘細胞を残せずにやがて死滅する。この例がヘルペスによって癌化した細胞をp53を働かせて癌細胞をアポトーシスさせることでヘルペスが大量に感染している癌細胞をherpesと癌細胞を同時に殺しているのです。したがって、チェックポイント機能の異常は遺伝情報の正確な伝達において大きな不具合を持つことを意味し、生物にとって重大な死をもたらす脅威となり実際に人の半分以上が癌で死ぬのです。実際にチェックポイント制御に関与するタンパク質群の変異が起きて、その変異を修正できなくなると癌死につながるのです。現代の癌治療は癌の原因はヘルペスであることを認めないでいかなるがん治療も人殺し医療で終わるのです。延命治療は延命できる期間が長ければ長いほど医薬業界だけが無限大のお金がもうかるだけです。悲しいですね。癌を治せるロイアルレイモンド博士の「癌光治療」はここを読んでください。
細胞周期チェックポイントの分類は細胞周期チェックポイントは、すでに述べたようにそのチェックポイントは長い複雑な細胞周期のどの段階(ステージ)に存在するかによって四つに分類され、これまでに (1) G1/Sチェックポイント、(2) S期チェックポイント、(3) G2/Mチェックポイント、(4) M期チェックポイントの4つがよく解析されている。これらはそれぞれ複数の、異なるチェックポイント制御関連分子によって制御されており、その細胞周期チェックポイント制御機構は極めて複雑であるのですが細胞周期チェックポイント制御機構の異常はヘルペスが自由自在に細胞のゲノムに自分のゲノムを組み込み遺伝子の部分的特異的組み換えを細胞周期が活動し始める細胞増殖とのための細胞分裂を行うときにやるからですという真実を世界中の癌学者は誰も気が付いていないからです。いやいや言い間違えました。私もとうとう嘘をついてしまいました。東大法学部を一番で入りかったのですが入り損ねたherpes脳炎持ちのヘルペス性神経炎で右目が失明して今もなおあらゆるヘルペス性疾患で悩んでいる世界一阿呆な半盲の医者が知っている真実を世界で最も優れた頭脳を持った医学部の教授たちが知らないと言うのは世界一のウソでした。御免なさい。改めて言いなおします。「癌の原因は癌ウイルスであるherpesウイルスが癌原遺伝子と癌抑制遺伝子の二つの遺伝子を突然変異させて起こったという真実を知っているくせに隠しているだけです。」に修正します。すいません。すいませんでした。この真実を故意に無視し続ける限り医薬業界のgenocideは永遠になくなりません。残念です。
G1/SチェックポイントはG1期からS期に移行する際のチェックポイントです。G1期DNAに損傷がないこと、これからのDNA複製のためのヌクレオチドなどが十分あること、細胞の大きさがチェックされる。多細胞生物では、増殖が許されているか(たとえば、サイトカインがあるか)、増殖が必要な細胞であるか、などもチェックされる。がん抑制遺伝子産物p53、RbとRbのホモログはこの制御を司っていると言われる。
この制御がDNA損傷などで活性化するとS期開始、すなわちDNA複製が阻害され、細胞はG1期にとどまる。酵母などで環境条件が良くない場合、または多細胞生物において細胞分裂が適当でない場合、G1停止が長く続くとG0期という休眠状態に入ることもある。G0期ではタンパク質合成が抑制され、細胞周期の進行に関わるタンパク質が一部分解される。
G1期からS期への移行

細胞が成長するG1期とDNAの複製が行われるS期の境界の細胞周期段階の現象について述べます。このG1期からS期への移行( G1/S transition)の段階は、細胞周期の完全性を保証する細胞周期チェックポイントによって制御されている。細胞はこの移行時に、環境要因や分子的なシグナル伝達による入力に基づいて、静止状態となる(G0期に移行する)か、分化するか、DNA修復を行うか、増殖を行うかを決定する。G1期からS期への移行時における、高度に調節されたチェックポイントの欠如や不適切な利用は、細胞の形質転換やがんなどの疾患状態を引き起こす可能性がでてきます。
この移行時にはサイクリンD–CDK4/6二量体はRbタンパク質をリン酸化し、転写因子E2Fを遊離させる。その後、E2FはG1期からS期への移行を駆動する。G1期からS期への移行は、DNAが損傷を受けている場合に細胞周期を停止することを目的として、癌抑制因子でもあり転写因子でもあるp53による調節を受ける。
この移行は細胞が分裂に従事する「回帰不能点」であり、その時点は酵母ではSTART、多細胞真核生物では制限点(R点)と呼ばれている。細胞がこの移行時点を通過した場合には、細胞周期の進行は分裂促進因子に依存せず、G1/S期の転写のポジティブフィードバックループによって進行が継続される。このポジティブフィードバックループにはG1期サイクリンとE2Fの蓄積が関係している。
細胞周期の概要は細胞周期は、細胞の成長と2つの娘細胞への分割をもたらす一連の順序立ったイベントからなる過程である。生み出された2つの娘細胞がこの周期を繰り返すため、細胞周期は直線的過程というよりはサイクルである。この過程は、細胞が成長しDNAのコピーを合成する間期と、細胞がDNAを分離して2つの新たな娘細胞へと分割される有糸分裂(M)期という主に2つの段階からなる。間期はさらにG1期、S期、G2期の三つに分けられ、M期は有糸分裂と細胞質分裂の二つに分けられる。細胞質分裂後のG1期の間、細胞は細胞環境の成長因子を監視しながら成長し、閾値となるサイズ(細胞種に特有のrRNAや総タンパク質量)を越えると、S期への進行を開始する。S期の間、細胞はM期のDNAを二つに分離するために重要な中心体や微小管形成中心の複製も行う。DNA合成の完了後、細胞はG2期に移行し、有糸分裂に備えて成長を続ける。有糸分裂は前期、中期、後期、終期といったサブ段階から構成される。有糸分裂中は、DNAは染色体へと凝縮され、紡錘体によって整列されて分離される。複製されたDNAがそれぞれ細胞の反対側の極へ分離されると、細胞質分裂の過程で細胞質は2つへ分割され、2つの娘細胞が形成される。
細胞周期の調節は生体内の大部分の過程と同様、腫瘍の形成につながる変異細胞の形成や無制御な細胞分裂を防ぐため、細胞周期は高度な調節を受けている。細胞周期の制御は生化学的な基盤を持ち、成熟促進因子)(MPF)のタンパク質が一連のチェックポイントに基づいてある段階から次の段階への移行を制御している。MPFはサイクリンとサイクリン依存性キナーゼ(Cdk)からなるタンパク質二量体で、細胞周期のさまざまな時点で結合して細胞周期の進行を制御する。サイクリンがCdkに結合すると、Cdk活性化されて他のタンパク質のセリン・スレオニン残基をリン酸化し、活性化や分解を引き起こすことで細胞周期の移行を可能にする。CdkはCDKと同じです。
G1期からの移行はG1期の中盤から終盤にかけて、Cdk4/6に結合したサイクリンDはS期サイクリン-Cdk複合体の構成要素の発現を活性化するが、S期サイクリンがG1期のうちに活性化されることは望ましくない。そのため、阻害因子であるSlc-1がS期サイクリン-Cdk二量体(複合体)と相互作用し、S期への移行の準備が整うまで不活性状態に維持している。細胞が成長してDNAを合成する準備が整うと、G1期サイクリン-Cdk複合体はS期サイクリン阻害因子をリン酸化し、ユビキチン化を引き起こす。ユビキチン化とはタンパク質の翻訳後修飾の一つであるユビキチン化は,細胞内タンパク質の選択的分解,DNA 修復,細胞周期,シグナル伝達,免疫など,多くの細胞内プロセスの制御に関与しています。そのため,ユビキチン化に関連した細胞機能調節システムの全貌解明は,基礎生物学的な観点だけでなく,医療への応用においても極めて重要です。ユビキチン化は,複数のユビキチン化関連酵素間でユビキチンを受け渡しながら進行します。これらの酵素の中で,ユビキチン連結酵素(E3)は,ユビキチン化すべき標的タンパク質に最終的にユビキチンを受け渡す酵素です。ユビキチン分子は,タンパク質の翻訳後修飾に使われる.その名のとおりユビキタスに発現し,ヒトにおいては少なくとも5000の標的基質が存在するとされる.また,ユビキチン化を誘導する酵素は,ヒトでは600以上とされることから,その複雑な制御メカニズムが容易に推測できる.ユビキチンはさまざまなコーディングシステムを持ち,目的によっては,分解系タグ,もしくは非分解系のタグとして機能する。
阻害因子のユビキチン化はSCF/プロテアソームによる分解のシグナルとなり、分解の結果S期サイクリン-Cdk複合体は遊離して活性化され、細胞はS期へ移行する。S期に入ると、サイクリン-Cdk複合体はDNA複製複合体のいくつかの因子をリン酸化し、複製複合体からの阻害タンパク質の離脱や複製開始を誘導する構成要素の活性化を引き起こすことでDNA複製を促進する。
Rbタンパク質とG1/S期の移行はG1期の中盤における他の重要な二量体は、Rbタンパク質(pRb)と転写因子E2Fから構成される。pRbがE2Fに結合している場合、E2Fは不活性状態である。サイクリンDが合成されてCdk4/6が活性化されると、サイクリン-Cdk複合体はpRbをリン酸化の標的とする。pRbはリン酸化に伴ってコンフォメーションが変化し、その結果E2Fは遊離して活性化され、遺伝子の上流に結合して遺伝子発現を開始する。具体的には、E2FはサイクリンE、Aなど他のサイクリンやDNA複製に必要な遺伝子の発現が駆動される。サイクリンEはpRBのさらなるリン酸化を誘導してE2Fをさらに活性化し、サイクリンEの発現をさらに促進する。サイクリンEはCdk2とも相互作用し、G1期からS期への進行を駆動する。
腫瘍形成におけるRbタンパク質の役割はRbの変異は目のがんの原因となる。pRbが変異して機能を喪失すると、E2Fを阻害することができなくなる。そのため、E2Fは常に活性化状態となり、G1期からS期への進行を駆動し続けることとなる。その結果、調節を受けない細胞成長と分裂によって目に腫瘍が形成される。
細胞周期チェックポイントは適切な細胞分裂を保証するため、細胞周期の進行を監視して異常時には進行を停止させる多数のチェックポイント(監視)が利用される。これらのチェックポイントには、4つのDNA損傷チェックポイント、G2期終盤の未複製DNAに対するチェックポイント、有糸分裂時の紡錘体チェックポイント、染色体分離チェックポイントなどがある。
調節因子としてのp53はG1期とS期には、細胞分裂に先立って適切な成長とDNA合成を保証する3つのDNA損傷チェックポイントが存在する。G1期の間、S期への移行の前、そしてS期の間に損傷したDNAはATMやATRの発現を引き起こす。ATM/ATRはその後、転写因子p53を安定化して活性化する。その結果、p53はp21CIPなどの遺伝子の上流領域に結合できるようになり、発現が誘導される。p21CIPは存在する全てのサイクリン-Cdk複合体に結合して阻害し、DNA損傷が修正されるまで細胞周期を停止させる。
DNA損傷チェックポイントにおける他の過程は4つのDNA損傷チェックポイントのうち、2つにはp53の活性化以外のDNA損傷監視過程が存在する。S期への移行時やS期の間、ATM/ATRはChk1/2を活性化することで、サイクリン-Cdk複合体の活性化を担うCdc25Aを阻害する。Chk1/2とは何でしょうか?Chk1/2のChk はcheckpoint kinase の英語の略でありチェックポイント・キナーゼと訳します。Chk1/2は Chk1とChk2の二つのキナーゼを意味します。DNAに傷害などの異常が感知された場合,ATM/ATRあるいはその他の因子によって発動され,これが下流のChk1/2やp53,Baxなどを活性化することによりDNA修復やアポトーシスなどが誘導され,遺伝情報が守られ癌化を防いでいるのです。ATMとはATRとは何ですか?ATM (Ataxia telangiectasia mutated)は,毛細血管拡張性運動失調症の原因遺伝子と同定され,ATR(ataxia-telangiectasia mutated related)は,Seckel症候群の原因遺伝子の1つである.ともに,グルタミンの1つ前のセリンまたはスレオニンを好んでリン酸化する.Seckel症候群とは希少先天性疾患のセッケル症候群(Seckel syndrome: SS)において、特定の遺伝子の変異によって細胞に生じるSeckel 症候群は,出生前から始まる成長障 害, 均整のとれた小人症, 小頭症, 後退した前額お よび下顎,湾曲した高い鼻をもつ鳥様顔貌,精神発 達遅滞を主徴とする疾患である。
活性化が阻害されることで、細胞は移行することができなくなる。S期に損傷したDNAを複製すると細胞やさらには生体全体にまで悪影響が及ぶ可能性があるため、これら2つのチェックポイントには追加の制御過程が存在する。
S期チェックポイントS期のDNA複製の速さを制御し、DNA複製に不具合が検知された場合、複製を遅らせる機構。DNA損傷ではヒトのATM蛋白質はこの制御に関与していると言われる。
G2/MチェックポイントG2期からM期に移行する際のチェックポイント。この制御がDNA損傷などで活性化するとM期開始が阻害され、細胞はG2期にとどまる。DNA損傷応答においては、ATR(ataxia-telangiectasia mutated related)がそれ自身かあるいは他の因子によって損傷を認識した後にリン酸化を受け活性化されると、ATRはChk1をリン酸化して活性化する。活性型Chk1はCdc25Aのリン酸化を促進し、Cdc25AによるCdc2の脱リン酸化を阻害するため、Cdc2は高リン酸化された不活性な状態に保たれ、M期に進行せずに細胞周期が停止する。また、活性化されたp53(遺伝子転写因子)は14-3-3s(シャペロン)を転写し、それがリン酸化Cdc25と結合し核外へ排出されるため、Cdc2が不活性なままになる。よってM期進行が抑制される。DNA損傷認識後のATRのリン酸化に関わっている因子は現時点でははっきりしていないが、ヒトがん抑制遺伝子産物BRCA1がその役割を担い、DNA損傷に応答したG2/Mチェックポイントの制御を司っているとも言われている。 DNA複製終了を待たずに、M期が開始する酵母変異株を考慮すると、監視(チェック)期間はS期からG2期にわたる比較的長い期間であると考えられる。
G2/M期DNA損傷チェックポイントG2/MチェックポイントはG2期とM期の間に位置する。G2/M期DNA損傷チェックポイントは真核生物における細胞周期の重要なチェックポイントであり、損傷した、もしくは不完全な複製が行われたDNAが十分に修復されるまで、有糸分裂の開始(G2期からM期への移行)が起こらないよう保証する機構である。G2/Mチェックポイントに欠陥を有する細胞において、DNA修復の完了を待たずにM期が開始された場合には、細胞分裂後にアポトーシスもしくは細胞死が引き起こされる。このチェックポイントは生化学的現象としてはM期サイクリン–CDK複合体の活性化であり、この複合体は紡錘体の組み立てを促進するタンパク質をリン酸化して有糸分裂中期への移行をもたらす。
サイクリンB/CDK1の活性サイクリンB/CDK1の細胞周期はサイクリン依存性キナーゼ(CDK)と呼ばれるタンパク質によって駆動され、CDKにはさまざまなチェックポイントでサイクリンと呼ばれる調節タンパク質と結合する。細胞周期のさまざまな地点において、特定のサイクリン-CDK複合体の活性化や不活性化が生じる。サイクリンB/CDK1の活性はG2/Mチェックポイントに特異的である。細胞が有糸分裂の開始に備える段階になると、サイクリンBが蓄積してサイクリン依存性キナーゼCDK1(酵母ではCdc2)の活性が増加する。Cdc2とは何か?ヒトではCDK1はCDC2遺伝子にコードされている。CDK1はサイクリンと複合体を形成してさまざまな標的基質をリン酸化し出芽酵母では75種類を超える基質が同定されている。Cdc2の活性は対応する活性化因子や阻害因子のリン酸化/脱リン酸化によってさらに調節されている。サイクリンB/Cdc2はホスファターゼCdc25を活性化し、その結果サイクリンB/Cdc2の阻害因子であるWee1)とMyt1が不活化される。Wee1とは,細胞周期を調節するタンパク質である。 細胞周期とは,細胞が成長し,分裂する過程のことで、細胞周期のブレーキとして機能し,細胞が損傷を受けたときに修復するための時間を生み出す役割を担っている。 WEE1が正常に機能しない場合,損傷を受けた細胞が修復されずに分裂を続けることがあり,細胞は死滅する。Myt1とは細胞周期の M期開始では Cdc2 の抑制キナーゼであるMyt1が不活性化されることが重要であるが、その分子機構は不明である。
Cdc25は複合体の活性部位からリン酸基を除去することで活性化を行うが、Wee1はチロシン残基をリン酸化することで複合体を不活性化する。このCdc25を介した活性化ループは、オーロラAキナーゼとBoraとの協調的相互作用によって間接的に増幅される。オーロラAキナーゼとはオーロラAキナーゼまたはオーロラキナーゼA(オーロラA、Aurora kinase A、Aurora A)は、ヒトではAURKA遺伝子にコードされる酵素である。ヒトの多くのがんで高頻度に過剰発現しているSerine/threonine-protein kinase 6という名称でも知られる。
G2期の間、Boraは蓄積してオーロラAと活性化複合体を形成する。その後、この複合体はPlk1の活性化を調節する。Plk1はWee1をリン酸化することでSCF複合体を介した分解の標的とするとともに、リン酸化によってCdc25を活性化し、こうした複合的作用によってCdc2を活性化する。Cdc2、Cdc25、Plk1、そしてサイクリンBの蓄積の複合的作用によってサイクリンB/Cdc2複合体は活性化され、有糸分裂の開始が促進される。
有糸分裂の開始は「全か無か」(all-or-none)型の応答である必要があるため、このポジティブフィードバックループに関与する多くのタンパク質はサイクリンB/Cdc2複合体の活性化を駆動する作用を果たす。サイクリン濃度が一定の最低活性化閾値に達すると、Cdc2の活性化は急速に進行する。この状態は活性が不活性化閾値(活性化閾値とは異なる値である)を下回るまで維持され、閾値を下回るとWee1とMyt1によるチロシンリン酸化によって急激に不活性化が進行する。DNA複製が完了していない場合、Cdc2の活性化閾値を上回るのに必要なサイクリン濃度はさらに上昇する。こうしたサイクリンB/Cdc2の双安定性とヒステリシスは、G2/Mチェックポイントの高度な調節を保証している。
DNA損傷応答経路上述したように、G2期にDNA損傷部位に局在するタンパク質は一連のシグナル伝達カスケードを開始し、サイクリンB/Cdc2の活性を介して有糸分裂の開始を制御する。サイクリンB/Cdc2活性に対する負の調節は有糸分裂の開始の遅れをもたらすが、この調節はS期以降に蓄積したDNA損傷の修復に重要であり、細胞分裂を継続するために必要である。
G2/Mチェックポイントで機能するタンパク質はもともと、酵母で放射線照射(radiation)に対する感受性の増大を示す変異体のスクリーニングから同定され、こうした変異体は”rad”変異体と命名された。これらの変異体では電離放射線照射や化学物質によるDNA損傷の修復が効率的に行われず、この経路に必要不可欠なタンパク質に変異が生じていることが明らかにされた。このチェックポイント経路の序盤のシグナル伝達に関与するタンパク質は、酵母ではRad3、脊椎動物ではATRと呼ばれるPI3K関連キナーゼファミリーのメンバーであり、このタンパク質はDNA損傷部位に局在すると考えられている。Rad3はRad26をリン酸化し、Rad26はチェックポイントの開始に必要である一方で、その維持には必要ではない。またRad3は、Rad1、Rad9、Hus1、Rad17など、その他のいくつかのタンパク質もリン酸化し、これらのタンパク質が存在しない場合にはチェックポイントは消失する。
Rad3の主要なエフェクターとなるキナーゼはChk1(checkpoint kinase 1)であり、このキナーゼはDNA損傷試薬に応答したG2/M期での停止に必要である。Chk1(checkpoint kinase 1)はM期サイクリンを不活性な状態に維持するキナーゼであり、S期から有糸分裂までの間にRad3によってリン酸化されることから、G2期での停止に特異的役割を有することが示唆されている。Chk1(checkpoint kinase 1)の過剰発現はDNA損傷に非依存的な細胞周期停止を引き起こす。さらに、Chk1の過剰発現はrad変異体の放射線感受性をレスキューする。これはChk1が有糸分裂の開始を遅らせることでDNA修復を可能にしているためであると思われる。
DNA損傷の存在によってATM経路もしくはATR経路が活性化され、それぞれChk2とChk1が活性化される。これらのキナーゼはCdc25とWee1の上流で作用し、サイクリンB/Cdc2複合体を調節する。Chk1とChk2はCdc25をリン酸化し、Cdc25の脱リン酸化活性を阻害するとともにユビキチン化による分解の標的とする。また、これらの経路はp53も刺激する。p53はCDK阻害因子p21や14-3-3タンパク質の機能を調節し、p21はCDK活性を阻害し14-3-3はCdc25を細胞質に隔離する。また、Chk1と14-3-3タンパク質は同様にWee1を正に調節することも示唆されている。Chk1によるWee1の高リン酸化は14-3-3の結合を引き起こし、Wee1を核内にとどめることでCdc2に対するリン酸化活性を高める。このように、Wee1とCdc25へのリン酸化はどちらもCdc2の活性化を阻害する。
ATM/ATR経路は、Wee1の安定性に寄与するPlkへの負の調節も引き起こす。Wee1とMyt1の安定化は、G2期での停止を保証し、DNA修復を可能にしている。
チェックポイントには複数の経路が関与しているため、Cdc25は細胞周期の遅れを引き起こす唯一の機構ではない。未複製のDNAや損傷DNAに応答して生じる、Chk1によるWee1に対する正の調節やCdc25に対する負の調節は、細胞周期G2期で強力に停止させる。Wee1量の増加とCdc25量の減少は、有糸分裂の駆動に必要なヒステリシスループのサイクリンB濃度閾値を高める役割を果たす。
チェックポイントの維持Rad3はChk1の活性化とG2期での停止の開始に必要であるが、DNA修復が十分に行われるようG2期での停止を維持するためには他のタンパク質が必要であると考えられている。こうしたタンパク質の1つがRad18)であり、Chk1がリン酸化されて活性化されている場合でもG2期での停止に必要である。このように、Chk1はチェックポイントの開始に、そしてRad18はチェックポイントの維持に必要である。また、Rad18はDNA修復に他の役割、具体的には染色体構造の維持に機能している。Rad18が存在しない場合、G2期での停止が他の手段で延長された場合でもDNAは修復されない。
こうしたG2期での停止の維持は、p53やp21によってさらに保たれている。p53もしくはp21が存在しない場合、放射線照射された細胞でも有糸分裂が開始されることが示されている。p21もしくは14-3-3蛋白が存在しない場合にはサイクリンB/Cdc2複合体を十分に阻害することができないため、G2/Mチェックポイントにはp53とp21による調節が必要である。p53の変異はチェックポイントに重大な欠陥をもたらすため、がん治療においても重要な意味を持っている。
チェックポイントの不活化Wee1とCdc25の双方が不活性化された場合には、G2/Mチェックポイントは消失する。Wee1の不在もしくは標的塩基残基であるTyr15が除去された場合にはCdc2活性の負の調節が取り除かれ、細胞は修復の完了を待たずに有糸分裂へ移行し、G2/Mチェックポイントは消失する。Cdc25が存在しない場合には細胞はG2期で停止し、G2/Mチェックポイントは活性化されることから、Wee1の活性化とCdc25の不活性化がチェックポイントの重要な調節段階となっていることが示唆される。またChk1の不活性化によって、DNA損傷の修復に関係なく、チェックポイントを乗り越えて有糸分裂への移行が促進される。一方で、活性化をもたらすリン酸化を除去するホスファターゼや活性化タンパク質のユビキチン化分解、独立した経路で有糸分裂を促進するチェックポイントアンタゴニストの作用機構など、チェックポイントの終結の正確な機構に関してはまだほとんど理解されていない。
癌では、CDK、サイクリン、p53など多くの細胞周期調節因子の発現に異常がみられる。より具体的には、これらは中心体に局在してG2/M期の移行に関与していることが示唆されており、これらのタンパク質を操作してがんの放射線療法や化学療法に対する感受性を高める研究が進められている。Chk1はDNA損傷に応答して機能するため、がんの薬剤標的として重要な意味を持っている。現在、G2/M期の移行の調節によって細胞毒性を発揮する化学療法薬の研究が行われており、G2/Mチェックポイントの阻害とチェックポイントでの停止の双方のアプローチが行われている。多くの治療法は、チェックポイントを不活性化することで過剰なDNA損傷を抱えたまま強制的に有糸分裂を開始させ、細胞死を誘導することに焦点を当てている。
M期チェックポイントM期(有糸分裂期)の途中にあるチェックポイントで、スピンドルチェックが行われる。M期の細胞では、G2期までのステップで複製された対を成す染色分体が、互いにセントロメア付近でコヒーシン複合体によって架橋結合し、また、このコヒーシンを切断するタンパク分解酵素セパラーゼがセキュリンと結合することで不活性化された状態で存在する。
有糸分裂過程の次のステップとして、細胞の両極から伸びる紡錘糸(微小管)が、それぞれの染色分体のキネトコア(セントロメアの一部)に結合する。一対の染色分体が対称になるよう、正しくかつ同時に、紡錘糸を介して細胞の両極に結合しているかどうかがチェックされる
分裂後期の染色分体の移動に際しては、ユビキチンリガーゼであるAPC/CがCdc20と結合して活性化することが必要となる。紡錘体が正しく形成されると、Cdc20の阻害タンパクであるMad2がCdc20との結合から外れ、APC/Cと結合する。活性化したAPC/Cによって、セキュリン蛋白がユビキチン化され、プロテアソーム依存的に分解されることでセパラーゼが活性化し、染色分体間を架橋するコヒーシン蛋白が切断される。これにより、染色分体は紡錘体極へと移動が可能となる。染色分体が両極から伸びた微小管と等しく結合していないうちは、オーロラキナーゼなどのスピンドルチェックポイントタンパクの監視によってAPC/Cの活性化が阻害され、染色分体の分離を抑制する。
紡錘体チェックポイント紡錘体チェックポイント(spindle checkpoint)は、有糸分裂または減数分裂において、複製された各染色体が紡錘体に正しく接着する(後期)まで染色体の分離を防ぐ、細胞周期チェックポイントである。スピンドルチェックポイント、紡錘体(スピンドル)形成チェックポイント(英: spindle assembly checkpoint、略称 SAC)、有糸分裂チェックポイント( mitotic checkpoint)とも呼ばれる。適切な染色体分離を行うためには、姉妹染色分体上の2つのキネトコアはそれぞれ反対側の紡錘体極と接着していなければならない。この接着パターンのときのみ、二つに分かれる各娘細胞が染色体の1つのコピーを受け取ることが保証される。このチェックポイントの生化学的特徴はM期サイクリン/CDK複合体による後期促進複合体の刺激であり、これによってサイクリンや姉妹染色分体をつなぎとめているタンパク質の分解が引き起こされる。
中期の開始は、染色体のキネトコアへの微小管の連結と、細胞の中央部への染色体の整列によって特徴づけられる。各染色分体にはキネトコアが存在し、姉妹染色分体の各キネトコアに結合した微小管はそれぞれ細胞の反対側の極から発したものである。これらの微小管は染色体を細胞の両側の極へ引っ張る力を発生させるが、姉妹染色分体間の接着がこの力に対抗する。
中期から後期への移行段階では、この姉妹染色分体間の接着は解消され、分離された染色分体は紡錘体微小管によって細胞の両側の極へと引っ張られる。染色分体は紡錘体極の物理的な移動によってもさらに分離される。未成熟な段階で染色分体の分離が起こると、染色体の誤分離と娘細胞での染色体の異数性が引き起こされる可能性がある。そのため、中期でのチェックポイントは、染色体が適切に接着するまで後期への移行を防ぐ役割を果たす。
細胞の同一性と適切な機能を維持するためには、各細胞分裂後に適切な染色体の数が維持される必要がある。染色体数が正常よりも少ないまたは多い娘細胞が形成されるエラー(異数性と呼ばれる)が生じた場合、最善の場合には細胞死が引き起こされるが、壊滅的な表現型が生じる可能性もある。
がん細胞では異数性は頻繁にみられる現象であり、これらの細胞では染色体分離に関与する装置や分離の適切な遂行を保証する機構に欠陥が存在することを示している。昔から多くのがんはそれを構成するそれぞれの細胞の染色体数が異なる異数性であることが知られ、がん細胞は、細胞分裂の度に染色体数が変動する染色体不安定性と呼ばれる .
ヒトのダウン症候群では親の減数分裂時の染色体分離の欠陥の結果、子の細胞は21番染色体の余剰コピーを有している。減数分裂時の欠陥によって余剰の21番染色体を持つ配偶子が形成され、この配偶子が受精後に21番染色体を3コピー持つ胚を形成する。
さまざまなタイプの遺伝学的研究により、紡錘体の脱重合、2つのセントロメアを持つ染色体の存在、セントロメアの異常な分離、出芽酵母(Saccharomyces cerevisiae)での紡錘体極の欠陥、キネトコアタンパク質の欠陥、セントロメアDNAの変異、有糸分裂で機能する分子モーターの欠陥など、多様な種類の欠陥がSACを活性化することが明らかにされている。
細胞分裂には遺伝子の複製とその遺伝子を娘細胞への分配の仕事があります。細胞のサイズが十分に大きくなり、または適切な刺激を受けて細胞が分裂の準備が整った際には、細胞は細胞周期の進行を開始する機構を活性化する。S期(細胞小器官合成期、DNAの複製期)には中心体を含む大部分の細胞小器官の複製が行われる。そのため、細胞分裂過程が終結したときには、各娘細胞は完全な細胞小器官のセットを受け取ることとなる。それと同時に、S期にはDNAの複製が非常に正確に行われる必要がある。DNA複製が完了すると、真核生物ではDNA分子は凝縮され、分裂期染色体が形成される。各分裂期染色体は2つの姉妹染色分体から構成され、姉妹染色分体間の接着が確立されている。各染色分体は完全なDNA分子であり、細胞の2つの極にそれぞれ1つずつ位置する中心体のいずれかと微小管を介して接着される。中心体と微小管によって形成されるこの構造はその形状から紡錘体と呼ばれており、染色体は2つの中心体の間に保持される。姉妹染色分体間の接着は後期に切り離され、各染色分体は微小管を介して接着している中心体へ向かって移動する。このようにして、細胞分裂過程の終結時に娘細胞が切り離された際には、それぞれが完全な染色分体のセットを受け取る。細胞分裂時の姉妹染色分体の正確な分配を担う機構は染色体分離(chromosome segregation)と呼ばれている。
染色体分離が正しく行われることを保証するため、細胞は正確で複雑な機構を発達させている。まず、細胞はDNA複製と二つの極における中心体の複製を協調的に行う必要があり、この過程に欠陥が生じると極が1つしかないとか、または多数の極を持つ紡錘体が形成される。こうした場合には染色体の分配のバランスが取れないため、異常な染色体分離が行われる。
有糸分裂による紡錘体への染色体の固定と染色体分離についてはS期の間に中心体は複製を行う。有糸分裂の開始時点で、双方の中心小体の長さは最大となり、さらなる様々な物質をリクルートして微小管核形成能力を増大させる。有糸分裂が進行すると、双方の中心体は分離して紡錘体を形成する。こうして紡錘体は微小管が発する2つの極を持つこととなる。微小管はタンパク質性の長い繊維で、非対称的な末端を持つ。一方の末端は(−)端と呼ばれ、比較的安定で中心体に近接している。もう一方の末端は(+)端と呼ばれ、伸長と短縮を繰り返しながら細胞の中心部で染色体を探索する。各姉妹染色分体にはセントロメアと呼ばれる特別な領域が存在し、その上にはキネトコアと呼ばれるタンパク質性の構造が組み立てられる。この構造は微小管の(+)を安定化することができる。そのため、細胞の中心部を探索している微小管が偶然にキネトコアと遭遇すると、キネトコアは微小管を捕捉し、染色体は姉妹染色分体の一方のキネトコアを介して紡錘体へ接着することとなる。染色体はキネトコアの紡錘体への接着に活発な役割を果たす。クロマチンにはRanのグアニンヌクレオチド交換因子(GEF)が結合しており、染色体近傍のRanはGDPの代わりにGTPの結合が促進される。Ran(ras-related nuclear protein)は低分子量Gタンパク質ファミリーの一つであり,GTP結合型とGDP結合型との間の変換によって,様々な細胞内反応を制御するスイッチとして機能する。活性化されたGTP結合型のRanは細胞質のタンパク質複合体からTPX2などの微小管安定化タンパク質を解離させ、染色体周辺で微小管の核形成と重合を誘導する。こうしたキネトコア由来の微小管は、outer kinetochoreのキネシンモータータンパク質ともに、紡錘体由来の微小管の側面との相互作用を促進する。こうした側面との接着は不安定であり、末端型の接着へと変換される必要がある。側面型の接着から末端型の接着への変換によって、微小管の(+)端の伸長と短縮は、染色体の適切な二方向型(bi-orientation)の接着を達成するために染色体を押したり引いたりする力へと変換される。姉妹染色分体間は接着されており、また双方のキネトコアは双方の染色分体上で背中合わせに位置しているため、一方のキネトコアが1つの中心体に接着されると、もう一方のキネトコアは反対側の極に位置する中心体へ向かって露出するようになる。そのため、ほとんどの場合2つ目のキネトコアは微小管を介して反対側の極の中心体と結合し、染色体は細胞分裂時の適切な分離が保証される基本的な二方向性配置(アンフィテリック英語でamphitelicとも呼ばれる)となる。時折、2つの姉妹キネトコアの1つが双方の極から発した微小管に同時に接着することがある。この配置はメロテリック(meroteric)と呼ばれ、紡錘体チェックポイントによっては検知されず、後期の間も中心部に取り残された染色体が形成され、染色体の異数性が生じる可能性がある。メロテリック型の配置は有糸分裂の初期には頻繁にみられるが、このタイプの配置はオーロラBによって検知されて取り除かれる。オーロラBはさまざまなタイプの腫瘍で過剰発現しており、抗がん剤開発の標的となっている。
有糸分裂中の姉妹染色分体間の接着については上述したように、姉妹染色分体はS期(DNA複製によって2つの同一なコピー(染色分体)が生成される時期)から後期まで結合したままである。後期には、姉妹染色分体は分離され、分裂中の細胞のそれぞれ反対側の極へ向かって移動する。酵母とアフリカツメガエルXenopus laevis卵抽出液での遺伝学的・生化学的研究により、姉妹染色分体間の接着において必要不可欠な役割を果たしている、多数のタンパク質からなる複合体が同定されている。この複合体はコヒーシン複合体として知られており、出芽酵母ではSmc1p、Smc3p、Scc1p(またはMcd1p)、Scc3pという少なくとも4つのサブユニットから構成されている。Smc1pとSmc3pは、高度に保存された染色体関連ATPアーゼのグループである、SMCタンパク質(Structural Maintenance of Chromosomes)と呼ばれるファミリーに属し、ヘテロ二量体(Smc1p/Smc3p)を形成する。Scc1pはRad21の出芽酵母ホモログであり、分裂酵母(Schizosaccaromyces pombe)でDNA修復に関与するタンパク質として最初に同定された。これら4つのタンパク質は酵母では必須であり、どれか1つにでも変異が生じると姉妹染色分体が早期に分離するようになる。酵母では、コヒーシンは染色体の腕に沿って優先的に結合し、セントロメアに近接して非常に豊富に存在することがクロマチン免疫沈降を用いた研究によって示されている。RAD21とは、ヒトではRAD21遺伝子にコードされるタンパク質である。RAD21(別名: Mcd1, Scc1, KIAA0078, NXP1, HR21)は必須遺伝子であり、出芽酵母からヒトまで全ての真核生物に進化的に保存されたDNA二本鎖切断修復タンパク質をコードする。RAD21タンパク質はコヒーシンの構造的構成要素であり、コヒーシンは姉妹染色分体間の接着(cohesion)に関与する高度に保存された複合体である。
ヘテロクロマチンの役割については古典的な細胞学的観察から姉妹染色分体はヘテロクロマチン領域でより強固に接着していることが示唆されており、ヘテロクロマチンの特別な構造または組成がコヒーシンのリクルートに有利に働いている可能性が示唆されている。事実、分裂酵母ではSwi6(HP1の分裂酵母ホモログ)はヒストンH3のメチル化されたリジン9番残基に結合し、コヒーシンのセントロメアリピートへの結合を促進する。ホモログとはホモログは相同遺伝子と訳し進化の過程で共通の祖先から由来する遺伝子を指し、構造や機能において類似性を持つことが多いです。このホモログという概念は、生物学の多くの分野で重要な意味を持ち、遺伝子の進化、機能、および生物間の関係性の解析に利用されます。
分裂酵母と脊椎動物の双方において、RNAi装置がヘテロクロマチンの確立を調節しており、この領域へのコヒーシンのリクルートを調節していることが示唆されている。RNAiとは、21~23bpの短鎖二本鎖RNA(siRNA : small interfering RNA)または長鎖二本鎖RNA(double-strand RNA; dsRNA)が、その標的遺伝子の転写産物 (mRNA)の相同部分を切断することにより、遺伝子の発現を抑制する現象です。
出芽酵母ではセントロメアに近接したヘテロクロマチン領域が存在しないにもかかわらず、機能的なセントロメアの存在によってそれに隣接する20–50 kbの領域でコヒーシンの結合の増加が誘導されることから、ヘテロクロマチン以外にもセントロメアでの接着の強化を保証する機構が存在しています。
これに関連して、ヒト細胞では有糸分裂中にOrc2(S期のDNA複製の開始に関与する複製起点認識複合体(ORC)に含まれるタンパク質の1つ)もキネトコアに局在している。酵母のOrc2は姉妹染色分体間の接着に関与することが示唆されており、その除去によってSACの活性化が誘導される。また、ORC複合体の他の構成要素(分裂酵母のOrc5など)も接着に関与することが観察されている。ORCタンパク質が関与する経路はコヒーシン経路に対して相加的に作用するようであるが、その大部分は未解明である。
接着の機能とその解消についてはセントロメアでの接着は紡錘体微小管による極方向への力に抵抗し、姉妹キネトコア間に張力を発生させる。この張力は、オーロラBが関与すると考えられている機構によって、微小管-キネトコア間の接着を安定化する。
事実、細胞内のコヒーシンレベルの低下は、姉妹染色分体の早期分離を引き起こすとともに、染色体の中期板への集合の欠陥や、オーロラBを含む染色体パッセンジャー複合体(chromosomal passenger complex)タンパク質の脱局在を引き起こす。提唱されているコヒーシン複合体の構造からは、この複合体が双方の姉妹染色分体を直接的に連結していることが示唆されている。この提唱構造では、コヒーシン複合体のSMC構成要素は構造的な役割を果たしているとされ、SMCヘテロ二量体はDNA結合タンパク質として機能し、そのコンフォメーションはATPによって調節されている可能性がある。しかしながら、Scc1pとScc3pが調節的な役割を果たしている可能性もある。
出芽酵母では、Pds1p(セキュリンとしても知られる)が姉妹染色分体間の接着を調節している。Pds1pはプロテアーゼEsp1p(セパラーゼまたはセパリン)に結合し、阻害を行う。後期の進行が開始されると、後期促進複合体(APC、APC/Cまたはサイクロソーム)がセキュリンを分解する。APC/CはRING型E3ユビキチンリガーゼであり、ユビキチンが付加されたE2ユビキチン結合酵素をリクルートする。セキュリンは、APC/Cの活性化サブユニットであるCdc20がAPC/Cのコアに結合している場合にのみ、APC/Cに認識される。セキュリン、Cdc20、E2酵素がすべてAPC/Cに結合すると、E2酵素はセキュリンをユビキチン化し、セキュリンは選択的に分解される。セキュリンの分解によってセパラーゼが放出され、セパラーゼは2つの姉妹染色分体を連結しているコヒーシンのリングを分解し、姉妹染色分体の分離が促進される。Polo/Cdc5キナーゼがScc1の切断部位に隣接するセリン残基をリン酸化し、このリン酸化が切断活性を促進することも示されている。
この装置は進化の過程を通じて保存されているが、脊椎動物では大部分のコヒーシン分子はAPC/Cの存在とは無関係に前期に放出される。この過程はポロ様キナーゼPLK1とオーロラBに依存している。しかし、ヒト細胞では少量のScc1は中期までセントロメアに結合したままであり、同程度の量が後期に切断されてセントロメアから消失する。一方で一部の実験からは、姉妹染色分体の腕部の接着は姉妹セントロメアの分離後に徐々に失われることで、姉妹染色分体が細胞の反対側の極へ向かって移動することも示されている。
染色体の腕部のコヒーシンの一部とセントロメアのコヒーシンはタンパク質シュゴシン(Sgo1)によって保護されることで、前期での放出を回避している。Sgo1がセントロメアの接着の保護因子として機能するためには、Pds1と同様に後期の初めに不活性化される必要がある。事実、脊椎動物ではPds1とSgo1はどちらもAPC/Cの基質である。
紡錘体チェックポイント(Spindle assembly checkpoint 略してSAC)とは何でしょうか?M 期に存在するチェックポイントであり、 Spindle assembly checkpoint (SAC)と呼ばれ、紡錘体微小管と各染色体上に存在するキネトコアが正確に結合するまで、細胞分裂を M 期の中期に停止させる。紡錘体チェックポイント(SAC)は、不適切な接着がなされているキネトコアから発せられるシグナルであり、すべての真核生物の間で保存されている。SAC(紡錘体チェックポイント)はCdc20を負に制御することで細胞周期を停止し、それによって後期促進複合体(APC)のポリユビキチン化活性の活性化を防ぐ。SACシグナルを担うタンパク質は、MCC(mitotic checkpoint complex)と呼ばれる複合体を構成する。MCCには、SACタンパク質、Mad2/Mad3(BubR1)、Bub3、そしてCdc20が含まれる。SAC(紡錘体チェックポイント)に関与する他のタンパク質には、Mad1、Bub1、Mps1(英語版)、オーロラBがある。高等真核生物では、さらにROD-ZW10複合体の構成要素、p31comet(、MAPK、CDK1–サイクリンB、Nek2(英語版)、Plk1などの調節因子が存在する。
チェックポイントの活性化についてはSAC(紡錘体チェックポイント)は不適切に連結されたキネトコアと紡錘体微小管の間の相互作用を監視しており、キネトコアが紡錘体へ正しく接着されるまで維持される。前中期の間、Cdc20とSACタンパク質は紡錘体への接着の前にキネトコアへ濃縮される。これらのタンパク質は、除去されて正しいキネトコア-微小管間接着がなされるまで、SAC(紡錘体チェックポイント)の活性化状態を維持する。1つでも未接着のキネトコアが存在すると、SACは維持される。微小管の(+)端の接着とキネトコア微小管の形成後、Mad1とMad2はキネトコアから除去される。チェックポイントの活性化の他の調節因子は、キネトコアの張力である。姉妹キネトコアがそれぞれ反対側の極に正しく接着しているときには、紡錘体の力によってキネトコアに張力が発生する。二方向型の接着がなされたキネトコアでの強い張力はキネトコアと微小管の組み立てを安定化するが、弱い張力は不安定化する作用がある。双方のキネトコアが一方の極に接着されるシンテリック(syntelic)型などの不適切な接着では、発生する張力の弱さによって不適切な接着は不安定化され、キネトコアが紡錘体へ正しい形で再接着を行うことが可能となる。染色体パッセンジャー複合体のオーロラB/Ipl1キナーゼが不適切なキネトコア接着の張力センサーとして機能し、微小管-キネトコア相互作用面に位置する、微小管切断KinI型キネシンMCAK、DASH複合体、Ndc80/Hec1(英語版)複合体の制御を通じて不適切な接着を不安定化する。オーロラB/Ipl1キナーゼは、1つのキネトコアが双方の極に同時に接着されているメロテリック(merotelic)型接着の修正にも重要である。メロテリック型接着は十分な張力を発生するため、SACによって検出されない。修正されない場合、染色分体のゆっくりとした移動速度のために染色体の誤分離が生じる可能性がある。
SAC(紡錘体チェックポイント)が活性化されると、MCCの活性の調節を介したAPCの阻害により、後期への移行は防がれる。MCCによるAPCの阻害機構はあまり解明されていないが、MCCはBubR1(Mad3)のKEN-boxモチーフを用いて偽基質としてAPCに結合すると考えられている。MCCの活性化と同時に、セントロメアタンパク質のCENP-E(英語版)もBubR1を活性化し、後期の進行を防ぐ。
MCCの形成についてはMCCはBub3と、Cdc20に結合したMad2、Mad3(BubR1)から構成される。Mad2とMad3はCdc20上の異なる部位に結合し、相乗的作用によってAPC/Cを阻害する。Bub3は、Mad3やBubR1のGLEBSモチーフと呼ばれるshort linear motifを介して結合を行う。MCCの形成のための結合の正確な順序はいまだ不明であるが、Mad2とCdc20が複合体を形成すると同時にBubR1、Bub3とCdc20も他の複合体を形成し、続いてこれら2つのサブ複合体が合体することでMCCが形成されている可能性がある[57]。ヒト細胞では、BubR1のCdc20への結合に先立ってMad2がCdc20に結合している必要があり、そのためMad-Cdc20サブ複合体がMCC形成の開始因子として機能している可能性がある。BubR1を欠失してもMad2-Cdc20のレベルはわずかに低下するのみである一方で、Mad2はBubR1-Bub3がCdc20に結合するために必要である。しかしながら、BubR1はチェックポイントの活性化には必要である。
MCCの形成機構は明らかでなく、キネトコア依存的な形成と非依存的形成という競合する仮説が存在する。キネトコア非依存的形成を支持する証拠としては、キネトコアの組み立ての核となるタンパク質が変異した細胞やSACが不活性化された細胞でもMCCが検出されることが挙げられる。このことはMCCはキネトコアへの局在がなくとも有糸分裂期に組み立てられることを示唆している。あるモデルでは、前中期の未接着キネトコアは、機能的なSAC(紡錘体チェックポイント)を介してAPCをキネトコアへリクルートすることによって、APCのMCCによる阻害に対する感受性を高めているとされる。さらに、Mad2とBubR1の欠失はキネトコア非依存的に有糸分裂のタイミングに影響を与えるが、他のSACタンパク質の欠失は有糸分裂の持続期間に影響を与えることなくSAC(紡錘体チェックポイント)の機能不全を引き起こすことが明らかにされた。SAC(紡錘体チェックポイント)は、第一段階ではMad2とBubR1がキネトコア非依存的に有糸分裂の持続期間を制御し、第二段階では他のSACタンパク質と同様に未接着のキネトコアが存在する場合に延長を行うという、二段階のタイマーとして機能している可能性がある。
一方で、現在主力となっているモデルは「Mad2鋳型モデル」であり、これはMCCの形成がMad2のキネトコアでのダイナミクスに依存するモデルである。Mad1は未接着のキネトコアに局在し、Mad2と強く結合する。Mad2とBubR1のキネトコアへの局在は、オーロラBにも依存している可能性がある[60]。オーロラBを持たない細胞は、染色体が微小管と接着していない場合でも中期での停止が起こらない[61]。Mad2には開いたコンフォメーション(O-Mad2)と閉じたコンフォメーション(C-Mad2)が存在する。未接着のキネトコアはまず Mad1/C-Mad2/p31comet複合体を結合し、未解明の機構によってp31cometを放出する。残ったMad1/C-Mad2複合体はO-Mad2をキネトコアへリクルートする。O-Mad2はコンフォメーション変化を起こし、C-Mad2となってMad1と結合する。このMad1/C-Mad2複合体はキネトコアへのさらなるO-Mad2のリクルートを担い、O-Mad2はC-Mad2へのコンフォメーション変化を起こしてCdc20に結合するという自己増幅反応が行われる。Mad1とCdc20はどちらも類似したMad2結合モチーフを持っているため、Cdc20への結合時にはC-Mad2へのコンフォメーション変化が起こる。このポジティブフィードバックループはp31cometによって負に調節されている。p31cometはMad1やCdc20に結合したC-Mad2に対して競合的に結合し、O-Mad2がC-Mad2へさらに結合していくことを防ぐ。下等真核生物にはp31cometが存在しないことを考えると、他の制御機構も存在する可能性がある。「鋳型モデル」という名称は、Mad1/C-Mad2がC-Mad2/Cdc20の形成の鋳型として機能することに由来している。こうしたC-Mad2/Cdc20複合体形成によるCdc20の隔離がSACの維持には必要不可欠である。
チェックポイントの不活性化については姉妹染色分体の正しい二方向型接着がなされた後にSAC(紡錘体チェックポイント)を不活性化する機構はいくつか存在する。微小管-キネトコア間の接着に伴って、ダイニン複合体によるstripping(引きはがし)機構によってSACタンパク質はキネトコアから遠くへ輸送される。引きはがされるタンパク質にはMad1、Mad2、Mps1、CENP-Fが含まれ、その後これらは紡錘体の極に再分布する。Strippingの過程は、未損傷の微小管構造と、微小管に沿ったダイニンの運動性に高度に依存している。p31cometはC-Mad2のポジティブフィードバックループの調節因子として機能するとともに、SACの不活性化因子としても作用する可能性がある。未接着のキネトコアは一時的にp31cometを不活性化するが、接着によって(おそらくリン酸化を介して)再活性化されてMad2の活性化を阻害する。他のSAC不活性化機構としては、Cdc20の非分解性ユビキチン化によるMad2-Cdc20複合体のエネルギー依存的な解離によるものがある。逆に、脱ユビキチン化酵素プロテクチンはSAC(紡錘体チェックポイント)の維持に必要である。未接着のキネトコアは継続的にMad2-Cdc20サブ複合体を再形成することによってチェックポイントを維持している。SACはAPCの活性化によるタンパク質分解によっても不活性化される可能性がある。サイクリンBのタンパク質分解とCDK1/サイクリンBの不活性化もSACの活性を阻害する。後期におけるMps1の分解は、姉妹染色分体間の接着の除去後のSAC(紡錘体チェックポイント)の再活性化を防ぐ。チェックポイントの不活性化後や細胞周期が正常に後期へ移行した場合、APCはMCCの活性の低下によって活性化される。このとき、酵素複合体は後期阻害因子であるセキュリンをポリユビキチン化する。セキュリンのユビキチン化と分解によって、セパラーゼと呼ばれるプロテアーゼが放出される。セパラーゼは姉妹染色分体を保持している接着分子を切断し、後期を活性化する。
キネトコアでの末端型の微小管接着がSAC(紡錘体チェックポイント)シグナル伝達の特定の段階を破壊する過程の説明として、新たな機構も提唱されている。未接着のキネトコアでは、MCC形成の第一段階はキナーゼMps1によるSpc105のリン酸化である。その後、リン酸化されたSpc105は下流のシグナル伝達タンパク質である、Bub1、Bub3、Mad1、Mad2、Mad3、Cdc20をリクルートすることができるようになる。未接着キネトコアでのMad1との結合はMad2のコンフォメーション変化を引き起こし、O-Mad2からC-Mad2へ変換される。Mad1に結合したC-Mad2はその後、他のO-Mad2分子と二量体化し、Cdc20周辺でのC-Mad2への変換を触媒する。このC-Mad2/Cdc20複合体は、他のMCCが形成されるようキネトコアにMad1とC-Mad2を残して解離する。MCCはそれぞれ2分子のCdc20を隔離し、APC/Cとの相互作用を防ぐことによってSAC(紡錘体チェックポイント)を維持している。Mps1によるSpc105のリン酸化はSACシグナル伝達経路の開始に必要かつ十分であるが、この段階はキネトコアへの微小管接着が存在しない場合にのみ起こる。内在性のMps1はNdc80のカルポニン相同ドメイン(CHドメイン)と相互作用することが示されており、Ndc80は染色体から離れたouter kinetochore領域に位置している。Mps1はouter kinetochoreにつながれているが、Ndc80の柔軟なヒンジ領域によってinner kinetochore内に局在しSpc105をリン酸化することができる。新たな提唱機構である機械的スイッチモデルでは、微小管のキネトコアへの末端型結合は2つの機構によってSACを不活性化するとされている。接着された微小管はNdc80のCHドメインとSpc105の間の距離を広げる。さらに、接着された微小管の周囲にリングを形成する、160のタンパク質からなる巨大複合体Dam1/DASH複合体が2つのタンパク質の間の障壁として作用する。この分離によってMps1とSpc105の間の相互作用が妨げられ、SACシグナル伝達経路は阻害される。
このモデルは動物を含むより高等な生物でのSAC(紡錘体チェックポイント)の調節には当てはまらないことに留意しておくことは重要である。出芽酵母のキネトコア構造には1本の微小管しか接着しないが、動物のキネトコアは多数の微小管の結合部位を含むはるかに複雑な網目構造をしている。そのため、SAC(紡錘体チェックポイント)の不活性化と後期への移行には、キネトコア結合部位のすべてに微小管が付着していることは必要とされない。すなわち、動物のキネトコアでは、SAC(紡錘体チェックポイント)が阻害されている間、微小管に接着した状態と接着していない状態が共存している。このモデルには、接着したキネトコアに結合したMps1が隣接する未接着キネトコアのSpc105をリン酸化するのを妨げるような障壁機構が含まれていない。さらに、酵母のDam1/DASH複合体に相当する複合体は動物細胞には見つかっていない。
SAC(紡錘体チェックポイント)の欠陥とがん。SAC(紡錘体チェックポイント)が適切に機能しない場合、染色体の誤分離や染色体の異数性、さらには腫瘍形成が生じる可能性がある。癌が生ずる形質転換はゲノムの完全性の維持が崩壊した時、特に染色体全体またはその大部分の領域で崩壊した時に生じ、加速される。事実、染色体の異数性はヒトの固形腫瘍の最も一般的な特徴であり、そのためSAC(紡錘体チェックポイント)は抗がん治療の標的となると考えられている。SAC(紡錘体チェックポイント)タンパク質とは紡錘体形成チェックポイント(SAC)は、細胞分裂に際して姉妹染色分体の動原体の不適切な接着を検出し、「後期で待機せよ」というシグナルを発することで染色体の分離を遅らせる監視機構である。通常、細胞周期のさまざまなチェックポイントは、高度に保存された冗長な機構を介してゲノムの完全性を管理しており、細胞の恒常性の維持や腫瘍形成の防止に重要な役割を果たしている。いくつかのSACタンパク質は、各細胞周期における適切な染色体分離を保証するために正と負の両方の調節因子として機能し、ゲノム不安定性とも呼ばれる染色体の不安定性を防いでいる。ゲノムの完全性は現在いくつかのレベルで評価が行われており、一部の腫瘍では塩基置換、挿入、欠失などの不安定性がみられる一方、大部分の腫瘍では染色体全体の増加または喪失がみられるのは何故でしょうか?染色体全体の増加または喪失が起こるのは2倍体の23対染色体が遺伝子複製の際に、23対の染色体の46本に分けるときや92本の姉妹染色分体にするときに染色体が十分に分けられていないときにヘルペスウイルスが無理やり染色体に侵入しようとするときに起こるのです。さらにキネトコアの接着、微小管の機能、姉妹染色分体間の接着などの異常がSAC(紡錘体チェックポイント)の欠陥がヘルペスウイルスによって生じて染色体の異数性が引き起こされる可能性がある。染色体の数が変異している状態を異数性といい、そのような. 変異を持つ個体を異数体(aneuploid)という。
ヘルペスウイルスによる有糸分裂調節タンパク質の変化が染色体の異数性を引き起こし、これががんでは高頻度で起こるという事実から、当初はこれらの遺伝子ががん組織で変異している可能性があると考えられていた。一部のがんでは、形質転換を引き起こす欠陥の原因となる遺伝子はよく特徴づけられている。多発性骨髄腫などの血液のがんでは、イムノグロブリン遺伝子の再編成にDNA切断が必要であるという特有の性質のために、細胞遺伝学的な異常はきわめて一般的である。一方、多発性骨髄腫ではMAD2などの主にSAC(紡錘体チェックポイント)で機能するタンパク質の欠陥も特徴づけられている。
また、大部分の固形腫瘍は主に染色体の異数体となっている。大腸がんに関しては、BUB1とBUBR1、そしてSTK15の増幅が、がんに至るゲノム不安定性への関与が示唆されている主要な調節因子である。乳がんでは、BRCA1遺伝子によって特徴づけられる遺伝性のがんは散発性がんよりも高いレベルのゲノム不安定性を示す。
Mad2やBubR1などのタンパク質の生理的レベルの変化は明らかに染色体の異数性や腫瘍形成と関係しており、このことは動物モデルを用いて実証されている。しかし、近年の研究ではそのシナリオより複雑なものであることが示されている。異数性は、組織でのSACの特定の構成要素のレベルの変化(低下または過剰発現のいずれか)によって腫瘍素因となる他の欠陥、すなわち、ヘルペスによるDNA損傷の増加、染色体再編成、細胞死の低下などが誘導されているときにのみ、高い腫瘍発生率をもたらす。また、SACの一部の構成要素は有糸分裂外での機能、Mad1は核内移行、Bub3は転写抑制、BubR1は細胞死、DNA損傷応答、老化、巨核球産生への関与が示唆されている。このことはすべて、腫瘍形成の増加が異数性だけではない他の欠陥とも関連していることを支持するものである。
BUB1やBUBR1のようにチェックポイントに影響を与えることが知られているがん関連変異は、実際のところは稀である。しかしながら、がんへの関与が示唆されているいくつかのタンパク質には紡錘体形成ネットワークとの関わりが存在する。p53などの主要ながん抑制因子もSACに役割を果たしている。ヒトのがんで最も一般的に変異している遺伝子であるp53がヘルペスによるがん抑制遺伝子変異により存在しない場合、細胞周期チェックポイントには大きな影響が生じる。p53はG1期チェックポイントで作用することが示されていたが、SAC(紡錘体チェックポイント)の調節にも同様に重要であるようである。また、がんの重要な面の1つとして、細胞死またはアポトーシスの阻害が挙げられる。IAP(inhibitor of apoptosis)ファミリーのメンバーであるサバイビンは、中心体近傍の紡錘体微小管と中期染色体のキネトコアに局在している。サバイビンはアポトーシスを阻害して腫瘍形成を促進するだけでなく、染色体分離や、より原初的な生物での役割と同様に有糸分裂の終盤段階の重要な調節因子であることが示唆されている。サバイビンとはサバイビン (survivin) は、inhibition of apoptosis (IAP) familyに属するヒト遺伝子である。サバイビンタンパクは、カスパーゼの活性化を阻害しアポトーシスを抑制する。サバイビンは癌細胞で高度に発現しているのに対し、完全に分化した細胞ではほぼ発現が見られない。癌細胞において、サバイビンの機能を破壊すると増殖が止まりアポトーシスが誘導されることから、サバイビンは癌治療において格好のターゲットである。サバイビンは、細胞周期においても高度に制御され、G2/M期でのみ発現する。
キネトコアの接着、微小管の機能、姉妹染色分体間の接着などのSAC(紡錘体チェックポイント)の他の側面も、欠陥が生じて異数性が引き起こされる可能性がある。がん細胞は、SAC(紡錘体チェックポイント)の回避によって多方向に分裂し、多極型の有糸分裂を引き起こすことが観察されている。多極型紡錘体での中期から後期への移行は不完全なセパラーゼサイクルを介して行われ、結果として染色体不分離が高頻度で生じ、がん細胞の異数性を増幅させるのです。
がんと遺伝的不安定性への関連については細胞周期チェックポイント制御の部分的破綻はがんの発生と進行(すなわち細胞の無制御な異常増殖)の大きなひとつの要因となります。主要ながん抑制遺伝子産物p53、Rb、BRCA1は細胞周期チェックポイント制御にも関与する。多くのがん抑制遺伝子産物はヒトのがんで頻繁に不活性化されており、多くのがんの原因であることが多い。細胞周期チェックポイントは正確な遺伝情報の伝達のための基本的な制御機構であり、その異常は遺伝的不安定性をもたらす。遺伝的不安定性とは何でしょうか?ゲノム不安定性または遺伝的不安定性( genome instability, genetic instability, genomic instability)は、特定の細胞系統のゲノムでみられる高頻度の変異を意味する。こうした変異には、核酸配列の変化、染色体再編成や染色体の異数性が含まれる。このような核酸配列の変異や染色体再編成や染色体の異数性はherpesは細胞分裂周期に染色体が46本にばらばらになる時やランダムに染色体のDNAに割り込んで部分的特異的組み換えをやるときにも遺伝子の塩基の配列に変異を起こして遺伝的に不安定な状態をもたらすのです。
多細胞生物ではゲノム不安定性は発がんに中心的な役割を果たし、ヒトでは筋萎縮性側索硬化症(ALS)などの神経変性疾患や神経筋疾患である筋強直性ジストロフィーの一因ともなっているのが筋繊維細胞に感染したヘルペスによるものです。DNA損傷部位や修復中のエラーを通過する際の不正確な転写は変異の原因となるため、ヘルペス感染のような外的要因による高頻度のDNA損傷はゲノム不安定性の一番大きな腹因となるのです。
エピジェネティックな変化または変異によるDNA修復遺伝子の発現の低下も、ゲノム不安定性の要因となる可能性も考えられます。代謝を原因とする内因性のDNA損傷も高頻度で生じており、ヒト細胞のゲノムでは代謝を原因とする内因性のDNA損傷は平均して1日あたり60,000回以上のため、DNA修復能力の低下はゲノム不安定性の重要な要因であり癌化の可能性が高いと言われていますが証明されてはいません。やはり100年前にロイアルレイモンド博士が実証したように癌は「癌ウイルス」であるherpesウイルスによって癌関連遺伝子が変異して変異が多くなればなるほど癌が拡大進行してあらゆる種類の転移も起こりやすくなるのです。
ゲノム不安定性の要因は、ヘルペスによる遺伝子の変異の積み重ねによるものです。DNA損傷部位や不完全なDNA修復中の不正確な転写は変異の原因となるため、ヘルペスによる外的要因が高頻度のDNA損傷はゲノム不安定性の一因となる。エピジェネティックな変化または変異によるDNA修復遺伝子の発現の低下も、ゲノム不安定性の要因となる可能性がある。エピジェネティックな変化とは遺伝子のオン、オフを制御するためにDNAに起こる化学的な修飾となります。 これらの修飾はDNAに対して起こるものの、DNAを構成している塩基配列を変えることはありません。 細胞内のDNA全体(ゲノム)の中で、遺伝子の活動(発現)を制御する修飾のすべてをまとめてエピゲノムと呼んでいます。
代謝を原因とする内因性のDNA損傷も高頻度で生じている(ヒト細胞のゲノムでは平均して1日あたり60,000回以上)ため、DNA修復能力の低下はゲノム不安定性の重要な要因である可能性が高い。代謝とは何でしょうか?代謝とは、生体内で起こる化学反応の総称で、生命活動そのものを指します。体内の成分を合成したり、エネルギーを産生したり、不要なものを排泄したりする一連のシステムです。
代謝とは何でしょうか?代謝には、新陳代謝と基礎代謝の2つの側面があります。新陳代謝は物質の合成と分解であり、基礎代謝はエネルギーの出入力でありエネルギー代謝ともいわれます。代謝を上げることで、免疫力や体温が上がり、生命維持機能が活発化します。また、老廃物の排出や細胞の更新、自律神経の調整にも効果があります。代謝が良い人の特徴としては、①筋肉量が多い②運動をする習慣がある③栄養バランスの取れた食事をしている④睡眠を十分とっている。代謝が悪いと、エネルギーを活用できていない次のような状態となり、①体が冷えている②元気がない③倦怠感を感じている④易感染状態にある⑤傷が治りにくい⑥排泄がうまくできない⑦体重が増えやすい⑧便秘気味である。
遺伝的不安定性は多くのがん細胞の主要な特徴であるのはヘルペスウイルスによる感染細胞のゲノムに自由自在にヘルペス自身のゲノムを組み入れ細胞の遺伝子を組み替えてしまい遺伝子の突然変異の積み重ねたために遺伝的不安定性が起こるのです。遺伝的不安定性とは、一般的には細胞が放射線に曝露された際に起こる初期の影響が残り、遺伝子の安定性が欠如された状態を指しますがほ広い意味での遺伝的不安定性と関連する概念には、①ゲノム不安定性は遺伝子が突然変異を起こしやすくなる現象で、がん化の本質の一つと考えられています。細胞分裂の際にゲノムをミスなく娘細胞に伝えていくシステムががん細胞ではヘルペスによって破綻していることが原因です。②染色体不安定性は癌や先天性疾患において染色体の数の異常、欠失、転座などが広範囲に見られる染色体の構造異常です。③遺伝性疾患は生殖細胞系列(精子や卵子)の遺伝子の異常によって起きる病気です。遺伝子の異常は偶発的に生じたり、親から遺伝したりすることもあります。正常な細胞分裂を保障するために、G1/S期など重要なところで細胞周期チェックポイントは、DNA未複製チェックポイント、紡錘体集合チェックポイント、染色体分離チェックポイント、DNA損傷チェックポイントから成り立っているのです。
DNA未複製チェックポイン トについてDNA未複製チェックポイントは、DNAの複製が完了して分裂期へと進む準備が整っているかを監視している。DNAの複製が未完了であると、ATR-Chk1依存的にM期への移行に必要なCdk/サイクリン複合体の活性化を阻害し、細胞 周期を停止させるのです。
紡錘体集合チェックポイントについて紡錘体集合チェックポイントは、M期後期で、紡錘体の形成が正常で分裂期の後期に移行できる状況かをチェックしている。紡錘体の形成に失敗していると、Mad2が微小管と結合していない動原体依存的に活性化され、分裂後期開始に必要なCdc20の活性を阻害し、染色体の分裂を停止する。
染色体分離チェックポイントについて染色体分離チェックポイントは、M期終期に、正常な染色体分配がなされたかをチェックしている。染色体分配に失敗していると、Cdc2/Cyclin B複合体が活性を失わず、細胞質分裂に移れないのです。
DNA損傷チェックポイントについてDNA損傷チェックポイントは、G1期、G1/S期、S期、G2/M期で働き、DNAに損傷も変異もない正常なDNA合成を保障している。この機構は、ATM/ATRがそれ自身によってか、あるいはその他の因子によって、DNA損傷を認識することによって活性化され、これが下流のChk1/2やp53、Baxなどを活性化することによって、DNA修復やアポトーシス、老化などによって、望まれない遺伝情報の喪失や細胞のがん化を防いでいるのです。
原発性の一個の癌細胞が増殖分裂するたびに周辺の細胞にヘルペスが感染すると次次とあらたに感染した細胞のゲノムに侵入して細胞分裂のたびごとに何百個、何千個とビリオンが増えていき新しい細胞の46本の染色体に潜伏感染しながら細胞のゲノムを突然変異細胞のDNA修復機構は変異が多すぎて細胞の遺伝子不安定性が増加して800もある癌関連遺伝子の突然変異が蓄積していきます。蓄積が多くなればなるほど癌の悪性度が増えてがん細胞の増殖性も勢いがついてヘルペスはますます癌細胞の癌関連遺伝子の突然変異を積み上げ増殖勢いがいや正にとめどなくいや増していきます。ヘルペスウイルスは一言でいうと遺伝子だけを持って感染細胞から増殖のために製造工場も何一つ自分を増やす材料は何一つなくエネルギーも一切合切、感染細胞に依存せざるを得ないのです。ヘルペスは何も癌を作るために細胞に感染しているのではないのです。自分の子孫となるビリオンを最大限増やすためです。
左図は人の臓器別の癌の各々に癌関連遺伝子がいくつ存在しているかがを示されています。この癌関連遺伝子臓器別の癌関連遺伝子が多ければ多いほどさらにその癌関連遺伝子をヘルペスが癌化する数が多ければ多いほどその臓器の癌細胞の悪性度が増えてさらにヘルペスも莫大な数になって遠隔転移もあちこちに見られるようになるのです。間違った癌標準医療は免疫を抑えるだけですから増えたヘルペスウイルスが自由神経終末ポリモーダル受容体に感染してしまい痛みにこらえきれずに終末期医療や緩和医療となりこの世とおさらばとなるのです。自由神経終末ポリモーダル受容体は何でしょうか?

左図にヘルペスによる組織損傷により生じる炎症第 1期の生体反応組織損傷により図のような炎症メディエーターがポリモーダル受容器を刺激する。ポリモーダル受容器は中枢に情報を伝えるだけでなく末梢性にサブスタンス Pやカルシトニン遺伝子関連ペプチド(C G R P)などを放出し、これらがまた炎症反応に関与するのです。因みに癌死する直前に全身に痛みが出るのは何故でしょうか?ガン最終末期には現代のがん医療は最大限免疫を抑制する間違った医療ですから最大限増えすぎたヘルペスはあらゆる細胞にherpesが感染してしまい、とりわけ左上の図にある人体のほとんどすべての組織に存在するポリモーダル受容器末端の細胞のほとんどに感染してしまうと全身の痛みに耐えられなくなり痛みを軽減するために麻薬を投与する緩和医療が行われて快楽の中で命が果ててしまうのです。
ヘルペスウイルスによる致死性の癌の増殖の勢いを示しかつ最も勢いのある悪性度の度合いとか進行性の強度や予後の不良性の度合いの順位を決定する最も確実な指標は何でしょうか?800個にも達する癌関連遺伝子の癌化遺伝子の数が癌の予後を決定します。従って各臓器癌の癌関連遺伝子の癌化の数を比較すれば致死性の癌の順位の予想が可能となります。実際の癌死の数と一致します。
圧倒的な第一位の死因は肺がんで年間に159万人がなくなっています。下図に示されているように肺がんの癌化の癌関連遺伝子の蓄積は非小細胞がんで147個です。小細胞癌の癌関連遺伝子は163個です。
第二位の死因は肝臓がんであり肝細胞癌の癌化の癌関連遺伝子の蓄積は39個です。
第三位の死因は胃がんであり胃細胞癌の癌化の癌関連遺伝子の蓄積は53個です。

第四位は結腸直腸がんで結腸直腸がんの癌化の癌関連遺伝子の蓄積は66個でした。第五位の乳がんでは52万人がなくなり癌化の癌関連遺伝子の蓄積は33個でした。第六位は食道がんの40万人で癌化の癌関連遺伝子の蓄積は食道の腺癌が57個で、食道扁平上皮癌は79個でした。他の癌細胞の癌化の癌関連遺伝子の蓄積数は上の図を見てください。他の臓器のヘルペスが癌化させる癌関連遺伝子の蓄積数は多ければ多いほど悪性度が高くなるので転移癌も多くなり致死率も増えるのです。ヘルペスが多ければ多いほど遠隔転移の数や量も増大するだけではなく悪液質(カヘキシア)よって栄養不良になりやすく。死期が早まります。
ヘルペスウイルスが最初に800個もある癌関連遺伝子の2種類の癌原遺伝子と癌抑制遺伝子のそれぞれを一個ずつ突然変異を起こしてしまうと一個の癌細胞が生まれます。この一個の癌細胞が分裂するときに一個のヘルペスウイルスが何百個も増殖によって金背うする正常な細細胞に感染して新たな仲間の癌細胞を生み出します。最初の一個の癌細胞を生み出したヘルペスはその癌細胞がまだヘルペスの増殖に利用できる限り免疫が上がれば細胞のゲノムに潜伏感染してその癌関連遺伝子の2種類の癌原遺伝子と癌抑制遺伝子を癌化させ続けます。このような癌を作りながら新新たに感染するサイクルを繰り返してヘルペスの増殖と癌細胞の増殖を細胞分裂と免疫が落ちる機会を狙って時間をかけて上述したサイクル策を繰り返して800個もある癌関連遺伝子をヘルペスが癌化させてがんは徐々に進行していくのです。癌を引き起こすのは毎日の生活の中で出会う活性酸素でもなく放射能でもなく、また紫外線でもないのです。
ヘルペスによって起こされる癌は血液癌以外の固形癌である上皮癌や肉腫になるのに時間がかかるのは何故でしょうか?人のタンパク質を作る遺伝子は一個の細胞に23500個あります。遺伝子はタンパク質の設計図です。この設計図の指令をタンパク質に変えるためにはタンパク質ではないmRNAを作るための遺伝子に含まれています。このエキソンを繋ぎ合わせてmRNAが蛋白の元になります。更にエキソンとエキソンンの間にイントロンがあります。イントロンとはmRNAのスプライシングによってmRNAから除去される塩基配列部分で蛋白の設計図にならない非コーディング領域で、遺伝情報の調節や多様性に関わっています。一方エクソンの役割とはアミノ酸に翻訳されコーディング領域とアミノ酸に翻訳されない非翻訳領域 (UTR)の二つがあります。コーディング領域はエキソンの塩基配列のうち、アミノ酸に翻訳される塩基配列領域を指す。非翻訳領域 (UTR)は 翻訳されないエクソンの塩基配列部分で、mRNAの安定性や翻訳の効率を調節します。
このようにゲノムには蛋白の設計図がない部分がゲノムの75%を占めエキソンはゲノムの2%を占めるだけでエキソンの内タンパク質に翻訳される正味のコード領域はさらに少ないのです。コード領域とは遺伝子のコーディング領域とも言われはずばりタンパク質に翻訳される領域を指す。つまりコーディング領域は成熟mRNA の5′ 非翻訳領域と3′ 非翻訳領域の間にある開始コドンと終止コドンに挟まれた正味のタンパク質に翻訳されるmRNA あるいはその鋳型となるDNA の領域を指すのです。この正味コード領域(タンパク質の設計図)全部合わせても、たったの1.2%に過ぎないのです。言い換えると実際には蛋白質の設計図はゲノムの1.2%だけで残りのゲノムの98.8%のゲノムの塩基の配列をヘルペスがゲノムに侵入して突然変異を起こしても癌のみならずあらゆる他の先天性の遺伝子病や後天性の遺伝子病は起こらないのです。しかも30億個の塩基から成り立っている人のDNAであるゲノムのうち人の遺伝子のトータルはゲノムの1.2%の23500個でありその内の合わせて800個だけが癌関連遺伝子の癌原遺伝子と癌抑制遺伝子ですから800前後だけが癌関連遺伝子は想像を絶する塩基から成り立っているゲノムもある癌ができるまでに突然変異を起こして癌化させ続けます。
ヘルペスウイルスのゲノムは15万塩基から成り立っています。ヘルペスウイルスのゲノムの総塩基数は15万個でヒト細胞のゲノムの総塩基数は30億個ですから15万÷30億=0.00005になります。いずれにしろヒト細胞のゲノムにヘルペスウイルスのゲノムを組み込むこむことは水が満タンの大きなどんぶりに水を一滴入れるだけの話しです。にもかかわらず何十年かけてストレスの多い人生をやりすぎるとストレスのために免疫が落ちて免疫では殺すことができないヘルペスが増えすぎて癌関連遺伝子が癌化することになるのです。癌は偶然になる病気になるのではなく一生をかけて自分で作る病気なのです。更にこれに追い打ちをかけるのが免疫を抑える薬を使って症状をとるだけのインチキ医学をお金儲けのためにやっているのが現代の世界中の医薬業界なのです。医薬業界の殺されないためには免疫を抑える薬を絶対使わないことです。とりわけステロイドは癌の原因であるヘルペスを無限に増殖させる人殺し薬ですからいかなる状況でも絶対に使用してはいけません。癌で用いる分子標的薬をはじめとする高価な抗体医薬品は免疫をおさえるだけの副作用の多い最悪の薬ですから絶対に使ってはなりません。癌のあらゆる現象はすべてヘルペスの増殖の結果なのです。癌細胞を抑える免疫療法はすべて無駄な治療です。癌を治す方法は二つあります。一つはロイアルレイモンド博士の「光癌療法」で癌の原因である癌ウイルスであるヘルペスウイルスを極めて安全で簡単に痛みもなく後遺症もなく金もかからない原因であるウイルスを殺すだけであらゆる転移癌も末期がんも100%完治してしまうのです。
二つ目はヘルペスはすべての人に感染しているので上述したように長い長い時間かけてじぶんで勝手に増やして癌になるのですからヘルペスを増殖分裂させないように抗ヘルペス剤を大量に飲み漢方煎じ薬で免疫をあげて新しい細胞に感染しないように努めれば私がすでに何人も完治させたようにヘルペスを殺すことはできませんから増やさないように心がければ治るのです。ただし末期がんで全身に遠隔転移してしまっている癌患者さんはヘルペスが全身に何百億超えてしまっている人は点滴の抗ヘルペス剤でないとヘルペスの増殖を抑えきれなくなります。とりわけバイタルオーガンである肺にヘルペスが作った肺癌患者で息が苦しいほど進行した人は極めて治すことはむつかしいのです。というのは酸素と二酸化酸素を入れ替えてくれる肺胞の細胞は幹細胞がないので肺胞の細胞を再生することがむつかしいのです。しかも肺には最も多い癌関連遺伝子がありますので肺細胞が人体で6億個もある肺細胞が癌化しやすいので癌の進行や悪性化や他の臓器への転移のしやすさが肺癌が最も速度が速くなります。癌の進行や悪性化や他の臓器への転移のしやすさは何が決めるのでしょうか? ヘルペスがその臓器に存在する癌関連遺伝子の癌原遺伝子と癌抑制遺伝子をどれだけ多く癌化することによって決まります。つまり人体には人体は,大きく 4種類の組織で成り立っています。組織とは,機能をもった細胞の集まりのことです。それらの組織が組み合わさり,器官や臓器になります。4種類の組織とは,上皮組織,神経組織,支持組織,筋組織のことです。私たちのからだは,約200種類,60兆個の細胞で構成されています。器官には骨格系、筋肉系、呼吸器系、循環器系、消化器系、神経系、腎泌尿器系、内分泌系の8つがあります。
因みにヒトゲノムの中では、タンパク質をコードするエキソン領域が1.5%なのに対し、イントロン領域は25%もあります。一部のイントロンは、遺伝子の発現を制御したり、翻訳を制御する低分子RNA配列を含むものがありますが、そのほとんどの役割はよく分かっていません。
一個のヘルペスが正常な細胞に感染して癌細胞に変えたりしている間に細胞が分裂するたび毎にherpesも何百、何千の数多くのヘルペスの子孫であるビリオンが生まれます。ヘルペスウイルスは遺伝子情報だけを持っているだけですから子孫を増やすための原料やエネルギーのすべてを細胞から略奪し、細胞を生きられなくすることもあり得ることを十分に理解してください。培養細胞では、5~6時間のエクリプス期(暗黒期)の後、感染ウイルスは対数的に増加し、14~16時間でピークに達するのです。暗黒期(eclipse period)とは、細胞にherpesウイルスを接種後、感染細胞内にherpesウイルス粒子が検出できなくなる期間の事で、エクリプス、陰性期、暗黒現象とも呼ばれる。暗黒期においてはウイルス粒子は脱殻を行い、裸の遺伝子だけとなりherpesウイルスタンパク質や核酸の合成を行っているのです。ヘルペスの子孫ウイルスであるビリオンの出現により再びherpesウイルス粒子の検出が可能となります。感染後にherpesの子孫ウイルスであるビリオンが細胞外に放出されるまでの期間を潜伏期と呼び、細胞膜表面で成熟して放出されるウイルスの暗黒期は潜伏期と一致します。
単純ヘルペスウイルスの細胞への感染からherpesウイルス粒子のビリオンの形成と成熟と放出までのプロセスをまとめておきます。1.吸着・侵入、脱穀。HSV(単純ヘルペスウイルス)はエンベロープ上のgCで細胞表面のプロテオグリカン(へパラン硫酸)に吸着し、gB、gDおよびgHによりエンベロープは細胞膜と融合し、カプシドは細胞内へ侵入する。核融合により細胞質内に侵入したヌクレオカプシドは核膜孔近傍に移動し、脱穀が起こり、核膜孔を通じてウイルスDNAが核内に放出される。2.ウイルスDNAの転写およびウイルス特異タンパクの合成・調節。ウイルスDNAは、細胞RNAポリメラーゼを利用して、転写される。ウイルス特異タンパクは、α―β―γの順に合成される(cascade regulation)。これらのタンパクは抗原性があり、それぞれimmediate-early antigen(前初期抗原)、early antigen(初期抗原)およびlate antigen(後期抗原)とよばれる。α遺伝子は5個知られており、その転写は、タンパク質合成阻害剤存在下でも起こる。αタンパクは2~4時間でピークに達する。HSVビリオン(感染性をもつ完全な粒子)のテグメント(エンベロープとヌクレオカプシドの間のタンパク)中のαTIV(VP16)はα遺伝子にトランスに作用し、α遺伝子の転写を促進する。αタンパクはいずれも調節機能を持ち、β遺伝子を活性化、βタンパクの合成を促進する。β遺伝子の転写は、αタンパクの合成が先行する必要があり、DNA合成阻害剤存在下でも起こる。βタンパクは感染後5~7時間でピークに達し、主に核酸代謝およびウイルスDNAの複製に関与する。β遺伝子にコードされるタンパクは、①DNAポリメラーゼなどDNA複製に必須なもの 、②チミジンキナーゼ(TK)、リボヌクレオチド・リダクターゼなどDNA合成に関与するが細胞の酵素でも代替できるものがある。①を欠く変異体は増殖できないが、②を欠く変異体は感染細胞が増殖分裂中の宿主細胞では増殖できるのです。γ遺伝子は、ビリオンを構成するγタンパクをコードしており、その発現にはαタンパクおよびβタンパクの合成とDNA複製が必要である。しかし、γタンパクには、gB、gDおよび主要カプシドタンパクなど感染初期および後期を通して合成されるものがある。3.ウイルスDNAの複製。ウイルスDNAは、TK(チミジンキナーゼ)とDNAポリメラーゼの遺伝子をもっている。ウイルスDNAは、感染3時間後より合成が始まり、9~12時間持続する。ビリオン内のDNAは線状であるが、感染細胞内では環状となり、ローリングサークル様式で複製される。4.宿主タンパクの合成阻害。ヘルペスウイルス感染により、感染により,宿主細胞の高分子合成は阻害されてしまいます。5.ウイルス粒子の形成・成熟、放出。カプシドを構成するタンパクは、細胞質で合成され、核に移動し、核内でカプシドを形成する。ローリングサークル様式で複製したDNAはゲノム単位に切断され、カプシドにパッケージされてヌクレオカプシドが形成される。核内のヌクレオカプシドは核膜をかぶり、一度核膜外腔に出た後、再び細胞質に侵入、テグメントを獲得し、ウイルス糖タンパクで修飾された細胞膜を最終的にエンベロープとして被り、空胞内に出芽、細胞外へ放出される。6.宿主細胞の変化。感染細胞には核膜封入体の形成と核膜の変化がみられる。感染初期の封入体はDNAに富み(HE染色で青色)、大きく(宿主細胞のクロマチンを核周辺部に圧排、核内大部分を占拠)、ウイルス抗原を含む(蛍光抗体染色陽性)。感染後期の封入体はDNAを失い(HE染色でピンク)、核辺縁部の宿主クロマチンとの間にhaloを生じ、ウイルス抗原を含まない(蛍光抗体染色陰性)。
網膜芽細胞腫(Retinoblastoma 略してRb)は通常5歳までの小児に発症する、発達中の網膜におこる悪性腫瘍である。RbはRB1遺伝子の両コピーに発癌の素因となる変異が生じた細胞から発症する。Rbは単発性または多発性の場合がある。Rb患児の約60%は片側性で診断時の平均月齢は24ヶ月、約40%は両側性で診断時の平均月齢は15ヶ月である。遺伝性RbとはRb易罹患性が常染色体優性で遺伝するものである。遺伝性Rbの患者においては、眼以外の腫瘍を発症するリスクも増加する。
RB1遺伝子とは何でしょうか?Rb遺伝子(Retinoblastoma Gene)とは癌抑制遺伝子の一つであり、網膜芽細胞腫の原因遺伝子として初めて発見された。細胞周期がS期へ移行するのを抑制しているほか、現在では多くの癌の発症に関与していることが分かっている。ヒトのRb遺伝子は染色体上の13q14.1-2に位置しており、Rbタンパク質(pRb)をコードしている。Rbタンパク質は928アミノ酸残基からなり、その機能はリン酸化によって制御されている。Rbタンパク質自身はDNA結合ドメインを有しておらず、E2Fなどの転写因子を介してプロモーターに結合する。
E2Fとは何でしょうか?E2Fは、高等真核生物で転写因子ファミリーをコードする遺伝子ファミリーである。そのうち3つはアクチベーターであり(E2F1)、E2F2、E2F3a)、他の6つはリプレッサーである(E2F3b、E2F4、E2F5)、E2F6、E2F7)、E2F8)。哺乳類の細胞でこれらは全て、細胞周期の調節とDNA合成に関与している。E2Fは転写因子として、標的プロモーター配列のTTTCCCGCコンセンサス配列(とそのバリエーション)に結合する。E2Fは、高等真核生物で転写因子ファミリーをコードする遺伝子ファミリーである。
Rb遺伝子産物は細胞周期の調節に関与しており、G1期における細胞周期の回転を抑制する。細胞周期とは細胞分裂の過程における4つの一連の過程を指す。細胞分裂が一度生じてから次に分裂が起こるまでの間を細胞周期の一周期とし、G1期、S期(DNA複製期)、G2期およびM期(細胞分裂期)の4つの時期に分けられる。G1期はS期の準備を行うと同時に、さらに細胞周期を進めるかの選択を行う時期でもある。G1期の中には臨界点(動物細胞では制限点)と呼ばれる時期があり、ここを通過すると細胞周期はS期へと進行していくが、このポイントを越えずにG0期(休止期)に入って分裂が停止する場合もある。
転写因子であるE2Fの転写活性化ドメインにRbタンパク質が結合することによりE2Fの活性を抑制することで、Rbタンパク質の転写調節作用において重要な役割を果たしている。E2FとRbタンパク質との結合は細胞周期依存的であり、G1/S期およびG2/M期ではRbタンパク質がサイクリンD-CDK4/6の複合体の働きを受けて高度にリン酸化されているためE2Fと結合できない状態にあるが、G1期前期ではRbタンパク質は脱リン酸化されているためE2Fと結合可能であり、E2Fにより支配される遺伝子(サイクリンA、サイクリンE、CDK2など)の転写を抑制する。
RB1変異に関連するがんの高い発生率は、この腫瘍抑制遺伝子が重要であることを浮き彫りにしています。RB1の機能喪失は、小児における網膜芽細胞腫に関連します。RB1遺伝子によってコードされるタンパク質であるpRbは、細胞周期の制御因子であり、RB経路の調節不全はヒトがんの大部分の形態において観察されます。 家族性がんに一般的に関連する腫瘍抑制因子の例を表1.2に示します。pRbのpはprotein(タンパク質)のpですからpRb はRbタンパク質の事です。

家族性がんに一般的に関連する腫瘍抑制因子は発癌しやすいメカニズムは何でしょうか?ヘテロ接合性の消失により 家族性がんが起こりやすくなるのです。ヘテロ接合性の消失とはLOH(loss of heterozygosity略してLOHで訳してヘテロ接合性の消失)と呼び,正常な対立遺伝子座(アリル)の欠失を意味するのです。 LOHが生じると,残りの正常なアリルに変異が誘発されやすいことが知られており,腫瘍抑制遺伝子の不活性化を引き起こす機構の1つとして重要なのです。このことは,網膜芽細胞腫家系(Rb)やLi-Fraumeni症候群(p53)などで腫瘍が多発するメカニズムと密接に関連しているのです。家族性癌や遺伝性癌が起こりやすくなるメカニズムをもっと詳しく説明しましょう。
がんは遺伝するのか?生殖細胞の遺伝子が癌化していれば遺伝子癌が起こります。
ほとんどのがんは遺伝しませんが、癌になりやすい変化が受け継がれる場合があります。

多くのがんは生まれてから後に遺伝子に生じた変化が原因であり、次の世代に遺伝することはありません。ただし、生まれながらにして癌に関わる遺伝子である癌抑制遺伝子に変化があると、次の世代にその変化が受け継がれる、すなわち「遺伝する」可能性もあります。
左図は遺伝性がんの発生の説明図です。細胞は「体細胞」と「生殖細胞」の二つに分けることができます。体細胞は筋肉や骨、神経や血液など体の多くのほとんどの部分を占める細胞です。これらの細胞に含まれる遺伝子に、生まれた後で変化が生じたとしても、次の世代に受け継がれることはありません。これに対し、生殖細胞は男性では精子、女性では卵子になる細胞です。そのため、もし生殖細胞に含まれる遺伝子に変化がある場合には、次の世代に受け継がれる可能性があります。
遺伝するがんと、遺伝しないがんとの違いは何でしょうか?対立遺伝子座(アリル)とLOHとは何でしょうか?ヒトの46本の染色体のうち半分のアリルと言われる対立遺伝子は父親から、半分のアリルと言われる対立遺伝子は母親から受け継ぎます。もし、卵子、または精子がもつどちらかの遺伝子に変化があると、子どもはその変化を受け継ぐ可能性があります。
ただし、どちらか片方の親から変化を受け継いだとしても、すぐに癌になるわけではありません。片方の遺伝子が変化によって機能しなくても、もう片方の遺伝子が正常であれば、その機能を補ってくれます。しかし、もし何かの原因でもう片方の遺伝子にも変化が起こると、両方の遺伝子が正常に働くことができなくなり、がんになる確率が高くなるのです。この現象をLOH(loss of heterozygosity訳してヘテロ接合性の消失)と言います。すなわち対立遺伝子座(アリル)の欠失を意味するので、 LOHが生じると,残りの正常な対立遺伝子座(アリル)である腫瘍抑制遺伝子の不活性化を引き起こす変異が誘発されやすいことが知られており,癌になる確率が高くなるのです。例えば,変異遺伝子がRbであると網膜芽細胞腫家系や変異遺伝子がp53であればLi-Fraumeni症候群などの腫瘍が多発するメカニズムはLOH(loss of heterozygosity訳してヘテロ接合性の消失)によるのです。

左図はがん遺伝子の変化と遺伝性の乳がんについての説明図です。乳がんの約1割は遺伝性のがんといわれています。遺伝性の乳がんを発症した人では「BRCA1」、または「BRCA2」とよばれる遺伝子に変化が見つかることが多くあります。
この遺伝子からできるタンパク質はDNAに生じた傷を修復する働きがあります。そのため、この遺伝子がうまく働かなくなると、遺伝子の変化が取り除かれずに蓄積してしまい、がんを引き起こす原因になります。「BRCA1」、または「BRCA2」に変化がある人すべてががんを発症するわけではありませんが、変化がない人よりも、発症するリスクは高くなることがわかっています。遺伝性のがんを引き起こす原因となる遺伝子としては他にも、「家族性大腸腺腫」の原因となるAPCや、「網膜芽細胞腫」を引き起こすRBなどが知られています。遺伝性がんであるかどうかは、遺伝子検査によって判断する必要があります。
がんの遺伝子の変化が遺伝性であるかを調べるには。がん遺伝子パネル検査で調べるのは、がんの組織や細胞、血液から取り出したDNAなどです。もし遺伝子の変化が見つかったとしても、それが親から受け継いだ遺伝性のものであるか、または生まれてから起きた変化であるかは、がん遺伝子パネル検査の結果だけからは区別をすることができません。
がん遺伝子パネル検査とは何でしょうか?次世代シークエンサーという装置を使い「がん関連遺伝子」を一度に解析します。がん遺伝子パネル検査は、がんの発生に関わる複数の「がん関連遺伝子」の変化を一度に調べる検査です。次世代シークエンサーとよばれる機械を使った新技術が使われています。がん遺伝子パネル検査の目的は患者さんのがんの遺伝子を詳しく調べて、一人ひとりに合わせた治療につなぐことが出来るといわれますが現代のがん医療は免疫を一時的に抑えるだけで、逆にherpesがこっそり増えるだけですから治すことが出来ないうえに癌を生み出すのはヘルペスであり癌の進行や転移や悪液質などの癌に関わるあらゆる現象や症状はヘルペスによるものであることに気づいていないので故近藤誠先生の言うように長生きしたければ現代のがん医療はすべて避けた方が長生きできるのです。がん遺伝子パネル検査では、患者さんのがん組織や血液からDNAなどを取り出し、「がん関連遺伝子」に変化があるかどうかを解析します。検査の対象となる癌関連遺伝子のセットのことを「パネル」とよびます。パネルには通常、複数の遺伝子が含まれ、使用する検査によって調べる遺伝子の数や種類が異なる場合があります。検査のデメリットは遺伝子の変化が見つからない場合もあります。また、変化が見つかっても、今までに例の少ない変化などの場合には、適切な治療法が限られる場合もあります。これまでの研究結果から、がん遺伝子パネル検査により遺伝子の変化に基づいた治療につながる割合はおよそ10%とされています。
がんのホールマーク(特徴)とは何でしょうか?がん研究者は、DNA増幅およびシーケンシング技術における大きな進展とともに、腫瘍形成過程に生じる細胞および分子の変化ならびに遺伝的不安定性に関する詳細な情報を提供してきました。がんゲノム解析は、腫瘍形成開始、がん進行、転移および薬剤耐性に関連する特定の変異の同定および機能的分類を可能にしてきました。 大規模な研究によって、腫瘍形成過程(増殖、エピジェネティックな修飾、成長および代謝、アポトーシス、ならびに他のプロセス)に異常性が発揮されるさまざまな細胞プロセスが同定されました。同様に、慢性炎症、病的血管新生、および免疫系回避は、集合的にがん進行および転移を促進します。
HanahanおよびWeinbergの提唱するがんのホールマークは、腫瘍細胞が示す特徴的で補完的な能力を表し、新生物疾患の複雑さを概念的に説明するフレームワークを提供します(図1.5)。これらの組織化された原理に焦点を当てることは、新しい研究分野を探索し、がん患者のために新しい治療方針を考案できるように研究者を支援することにつながります。
がんとは体細胞遺伝子の変異の蓄積で生まれますがどのように変異の蓄積が起こるのでしょうか?具体的には800個もある癌関連遺伝子には2種類あります。一つは癌原遺伝子であり二つ目は癌増殖抑制遺伝子です。この二つともがヘルペスによって変異させられると一個の癌細胞が生まれます。800個もある癌関連遺伝子の二つが癌化しても増える勢いは正常細胞以下の増殖の勢いです。この癌細胞に長期に感染し続けている間にヘルペスはこの癌細胞が分裂するたびに免疫の状態に応じて大小はありますが増えていきます。増えたビリオンはこの癌細胞のゲノムに溶原感染して部位特異的遺伝子組み換えをやり偶々癌関連遺伝子の変異を起こすと癌関連遺伝子の変異が徐々に蓄積して悪性度を高めていくと増えるスピードが速くなっていきます。これは一個の細胞が転移して定着した癌細胞の転移巣で増えていくのも同じやり方で転移した癌細胞も癌関連遺伝子の変異が徐々に蓄積して悪性度を高めていくと増えるスピードが速くなっていきます。ところが転移巣に原発巣から増えた癌細胞が転移してくる数が多くなればなるほど転移巣でどんどん原発巣の癌細胞は転移巣でも増えて転移巣の癌細胞塊を大きくしていくのです。
人および小児腫瘍の多様なアレイ(対立遺伝子)のゲノムのシーケンシング(塩基配列)研究により、さまざまなヒトの癌関連遺伝子変異である体細胞の変異の数およびタイプについての情報が得られました(図1.6) 。例えば、身体のさまざまな組織や細胞に生じる体細胞変異の数は、小児ラブドイド腫瘍に見られるわずか4変異から、小細胞肺がんに見られる163変異までに及びます。ラブドイド腫瘍には、腎臓に発生する腎ラブドイド腫瘍と、脳や脊髄に発生する非定型奇形腫様ラブドイド腫瘍(AT/RT)などがあります。腎ラブドイド腫瘍は、腎臓の正常な細胞が変異して制御不能な細胞成長を始めることで発生する悪性腫瘍です。主に小児で診断され、急速な成長や他の器官への転移が特徴です。非定型奇形腫様ラブドイド腫瘍(AT/RT)は、脳や脊髄の組織の中に悪性(がん)細胞ができる疾患です。小児ラブドイド腫瘍は2歳以下の乳幼児の脳や脊髄に発生し、小脳など脳の下部に発生することが多いです。中枢神経系に発生する極めて悪性度の高い腫瘍で、男児に多くみられます。小児ラブドイド腫瘍は中枢神経系が胎児であるときに完成するとき時にヘルペスに感染していくつかの臓器に感染したために2歳以下の乳幼児の脳や脊髄に発生するのです。発症部位によって症状が異なり、中枢神経の場合、頭痛や嘔吐、意識障害やけいれん、麻痺などの脳圧亢進症状が現れます。
乳房、結腸、脳、ならびに他の器官および組織に発生する一般的な固形がんでは、タンパク質産物を変化させるわずかな体細胞変異の平均数は33から66です。これらの変異の95%以上は、1塩基置換です。約5%の変異では、欠失、転座、または挿入が観察されます。正常細胞において生じる点変異(塩基の一つが変異する)の割合は、腫瘍細胞において生じる点変異の割合に反映されます。しかしながら、がん細胞では染色体変化の割合が上昇します。これらのヘルペスによって生ずる変異では、染色体の数、欠失、転座、遺伝子増幅、および他の異常において変化が生じます 。遺伝子増幅というのは遺伝子のコピー数増加であり同じ遺伝子が多くあることは癌細胞の特徴の一つです。といっても過言ではありません。
下図に癌の性質の10個のホールマーク(特徴証明書)を示します。①持続的な増殖シグナル②増殖抑制因子からの回避。③免疫からの回避④無制限の遺伝子複製能による不死化⑤細胞死に対する耐性⑥ゲノムの不安定性と変異⑦浸潤と転移の活性化⑧腫瘍を促進する炎症⑨血管新生の誘発⑩調節解除された細胞エネルギー産生。の10個です。これらの癌の10個の特質は実はヘルペスウイルスがもたらした特徴です。このホールマークと癌細胞を作り細胞がヘルペスに利用しつくされるまでの特色なのです。

①持続的な増殖シグナルは癌細胞が分裂するたびにヘルペスが何百匹のビリオンを生み出すたびにヘルペスが増殖して癌細胞のゲゲノムに侵入して癌関連遺伝子の癌原遺伝子の変異が徐々に蓄積して癌細胞の増殖性が増えるのです。
②増殖抑制因子からの回避は①と同じで癌関連遺伝子の癌増殖抑制因子の機能変異が重なり機能が増殖抑制の機能がなくなっていくのです。
③免疫からの回避は元来癌細胞は自己の細胞ですから遺伝子が少しばかり変異しても非自己と認識されないのみならず癌細簿に居座っているヘルペスウイルスが時に癌細胞外に出ても元来ヘルペスはワクチンができないので免疫は認識しにくいのです。
④無制限の遺伝子複製能による不死化⑤細胞死に対する耐性の二つは同じ事ですから一緒に説明します。確かに癌は細胞死(アポトーシス)が起こりにくくなっています。
細胞が分裂増殖するには自身のDNAを複製する必要がありますが、通常のDNAポリメラーゼを介した仕組みではDNA鎖の両端(テロメアDNA)が完全には複製されず、徐々に失われていきます。これを末端複製問題(end replication problem)といいます。テロメアはもともと染色体の末端を保護する役割を持っていますので、その短縮が限界に達しますと、DNA鎖の先端がむき出しになってしまいます。DNAの複製工場は操業停止となり、細胞はもはや分裂することが出来なくなります。細胞も老化する、というわけです。テロメアは細胞老化のいわば時限装置として働いており、これは、私たちの身体の中で、異常な増殖性を持った細胞ががん化するのを未然に防ぐ仕組みのひとつとなっています。がん細胞ではたいてい、テロメラーゼ(telomerase)と呼ばれるテロメア合成酵素が活性化しており、この酵素の働きによってテロメアが安定に維持されます。がん細胞が無限に分裂出来るのはこのためです。これは嘘です。
がん細胞の最大の特徴の一つとして、何度でも細胞分裂を続けることができるという性質があります。この「がん細胞が無制限に細胞分裂できる能力」のことを専門用語では「細胞不死化能」といいます。一方、正常な細胞は、細胞分裂の回数に制限があり、無制限に細胞分裂を繰り返すことはできません。このように、がん細胞には不死化能があるのに対して、正常細胞には不死化能がないことが大きな違いであったため、約30年にわたり多くの研究者によって、がん細胞はどのようにして細胞不死化能を獲得するのかという研究が行われ、1997年に細胞不死化酵素としてテロメラーゼという分子が発見されました。その後も、世界中のがん研究者らは、細胞不死化能の有無に着目して、テロメラーゼを阻害する薬剤の開発を進めてきましたが、がん治療として十分に効果がある薬剤は開発できませんでした。失敗の原因はテロメラーゼもテロメアも癌の不死化と関係がないからです。それでは癌細胞が死化能を持つことができるのは何故でしょうか?テロメアの短縮そのものが実は直接の原因ではなく「テロメアの短縮」を異常事態として感知する細胞チェックポイントが正常に働いて細胞分裂が止まってしまうからです。テロメアの短縮そのものが細胞分裂を止めたわけではないのです。何故でしょうか?つまり細胞分裂がある回数に達した時点で分裂を止めとめることを細胞の老化でありこれは分裂のたびに不可逆的な変化であるテロメアの短縮であります。テロメアの反復配列が欠落してテロメアの短縮をDNAの損傷と認識した癌関連遺伝子の癌増殖抑制遺伝子の代表であるp53はDNAの損傷を持った細胞(癌細胞)を増殖させてはならないと判断してDNA損傷チェックポイントの機構が作動し始めるのです。DNA損傷チェックポイントの機構と同じくp21cp1を介して細胞周期を停止させて細胞分裂をさせないようにしているのです。本来はダメージを受けた細胞をアポトーシスで排除する代わりにおとなしく封じ込めておくのが「老化」であるから生体から見ると一種の防衛機構であるのですがこれが予期せぬ不都合を生じることがあるのです。それが発がんへの影響なのです。通常小さな癌が生じても周りの正常組織が抑制的に働きます。ところが老化した細胞ではこの能力が小さな癌に対して低下しているだけでなく老化細胞はMMPや上皮成長因子や炎症性サイトカインの様な分泌タンパク質などを過剰に産生してしまいます。この様な分泌タンパク質などの作用でむしろ癌の増殖や進展を促進させる傾向を持っているのです。元々は発がんを防ぐための手段であったテロメアの短縮による老化が逆に発がんをサポートしてしまうという皮肉な結果をもたらしてしまうのです。
というよりも癌原因はヘルペスですから癌の本質はたった一つ「ヘルペスが感染細胞の癌関連遺伝子の癌原遺伝子と癌増殖抑制遺伝子をヘヘルペスが変異させなければがんは絶対に起こらない。」ので100年前にロイアルレイモンドライフ博士が「光癌治療」で癌ウイルスであるヘルペスウイルスを殺し切って3か月でお治し切ったのは奇跡であったわけでもなくただ原因をなくしただけですから難解な癌理論も何も必要は無かったのです。彼の作った簡単な「光癌治療」装置が復活すれば癌という病気は風邪よりも治しやすいどうでもよい病に格下げできるのにつまらない理論と医療をはびこらせ金を儲ける罪びとである医者が多すぎます。残念です。
また、近年、世界中のがん研究者が参加する国際共同研究による、「がん全ゲノム解読」によりテロメラーゼが大変重要であることが改めて証明されることとなりました。例えば、C型肝炎関連肝臓がんなどでは、患者検体を用いた大規模ゲノム解析で、約7割の症例でテロメラーゼが発がん過程における最も重要な分子であることが報告されています。テロメラーゼは、もともと細胞不死化酵素として同定された分子であることから、これまでは、このテロメラーゼの役割は唯一、「細胞に不死化能を付与する」ものと理解されてきましたが、「テロメラーゼの細胞不死化能を阻害する」という考え方に基づくと、がん治療薬の開発が順調に進展しなかったことなどから、テロメラーゼは癌化とは関係がないと証明されています。テロメラーゼやテテロメアは本来は発がんを防ぐための手段であったのが「細胞を老化させるためにあるのです。これをテロメラーゼやテロメアを「発がん」と結びつけようと「癌医学会が」が仕組んだのですが失敗に終わったのです。
ところがテロメアの減少によって人の細胞の分裂回数をもたらして細胞の老化を促進させて癌が増殖できないようにして癌による短命を防ごうとしたのですが老化なのですが皮肉なことに老化が逆に癌を増殖する働きがあるのです。癌細胞の敵であることで有名な癌抑制遺伝子の代表であるp53の発現が亢進したマウスでは確かに発がん率は低下しますがその分、個体の老化が促進して短命になってしまうこともわかったのです。
更に老化に特異的なタンパク質であるSA-β-gal (Senescence-associated beta-galactosidase略してまたはSABGで訳して老化関連β-ガラクトシダーゼ)は老化した組織に増えます。SA-β-gal (Senescence-associated beta-galactosidase訳して老化関連β-ガラクトシダーゼ)とは老化細胞でのみβ-ガラクトシドの単糖への加水分解を触媒する加水分解酵素です。老化関連ベータガラクトシダーゼは、p16ᴵⁿᵏ⁴ᴬとともに、細胞老化のバイオマーカーです。ベータガラクトシダーゼアッセイをpH 6.0で行うと、老化状態の細胞のみが染色されます。この現象は、老化細胞に特異的な内因性リソソーム β-ガラクトシダーゼの過剰発現と蓄積によるものです。リソソーム β-ガラクトシダーゼは老化細胞と加齢細胞の最も広く使用されているバイオマーカーとなっています。老化のバイオマーカーはリソソーム β-ガラクトシダーゼです。
このSA-β-galという老化特異的なタンパク質は動脈硬化、関節炎、前立腺肥大、肝臓肥大のような老化性の組織に多く見られます。更に老化した組織内で認められるSA-β-gal要請の細胞では同時にp16ink4aも増えていることが多いのです。
p16ink4aとはすでに説明したのですがp16とはサイクリン依存性キナーゼ阻害2A (cyclin-dependent kinase inhibitor 2A) は、CDKN2Aとも言い、ヒトのCDKN2A(p16)遺伝子によりコード化されているがん抑制タンパク質である。p16は細胞周期の調整に重要な役割を果たしており、p16の変異は様々な癌、特にメラノーマの発生のリスクを高めている。なお、CDK(cyclin-dependent kinase)とはサイクリン依存性キナーゼの略である。上で説明したように細胞の老化と関連が深いのです。
⑥ゲノムの不安定性と変異はヘルペスウイルスが正常細胞に感染してゲノムの癌関連遺伝子を変異させかつゲノムのあちこちの塩基を変えたりしてゲノムの不安定性をもたらすのです。癌に関わるゲノムの不安定性の中で最も重大であるのは癌関連遺伝子である癌原遺伝子と癌抑制遺伝子の変異です。この癌関連遺伝子はすでに確認されているだけでも800以上もあり癌原遺伝子と癌抑制遺伝子のそれぞれの遺遺伝子のである特徴となる①遺伝子産物②オンコジーンとの対応③遺伝性の癌との関係④人染色体上の位置⑤遺伝子の名前の⑥癌発生における役割すべてが解明されています。①~⑥までについて詳しく説明しましょう。
①遺伝子産物。遺伝子産物とタンパク質であり癌細胞でない正常な細胞であれば正常な役割を果たすタンパク質が生まれます。ところが癌関連遺伝子である癌原遺伝子と癌抑制遺伝子の変異がヘルペスウイルスによって起こされて二つの遺伝子が癌遺伝子(オンコジーン)になると異常なたんぱく質が産生され発がんタンパク質となってしまうのです。
②オンコジーンとの対応。オンコジーン(oncogene)とは、「オンコジーン(oncogene)」は日本語に訳せば「がん遺伝子」ですがもっと正確に言えば「癌原遺伝子」です。「がん遺伝子」になる前の「すべての正常な細胞が持っている遺伝子という意味で癌原遺伝子なのです。」正常な細胞増殖のシグナル伝達を担うのですがヘルペスによって、癌原遺伝子がオンコジーン(oncogene)といわれる異常な「癌遺伝子」になると発癌をもたらすのです。更に癌になるためにはアンチオンコジーン(発癌 抑制遺伝子)といわれる遺伝子が変異しなければガンにはなりません。アンチオンコジーンとは発癌 (はつがん) 抑制遺伝子であり、癌遺伝子(オンコジーン)と同様に遺伝子の一つであるが、癌遺伝子(オンコジーン)とは逆に癌化を防ぐ作用があるのです。癌遺伝子(英語はオンコジーンですが日本語訳は癌原遺伝子とも訳す。)は正常細胞の遺伝子に由来するもので、潜在的にがん遺伝子になり得る性質を持つ遺伝子は「がん原遺伝子(proto-oncogene)」と呼ばれます。
がんの発生には、細胞の分裂や増殖に関わる遺伝子が変異する必要があります。がんに関連する遺伝子には、二種類あります。一つ目は細胞を増殖させる「癌細胞を増殖する遺遺伝子」であり「アクセルの役割をする遺伝子」ともいわれます。二つ目は細胞増殖を抑制する「がん抑制遺伝子」であり「ブレーキ役の遺伝子」ともいわれます。傷ついた遺伝子を「修復する遺伝子」でありがん化を防ぐ作用を持つ遺伝子なので「アンチオンコジーン(anti-oncogene)」や「発癌抑制遺伝子」ともいいます。p53、p16、PTENなどががん抑制遺伝子に含まれます。
レトロウイルスとは何でしょうか?因みに癌遺伝子(オンコジーン)はレトロウイルスが持っている癌原遺伝子を指すこともあります。レトロウイルスとはRNAウイルス類の中で逆転写酵素を持つ種類の総称。プラス鎖ゲノムの一本鎖RNA(ssRNA)から成る。単にレトロウイルスと呼ばれることも多い。逆転写酵素を持つものは、必ずしもRNAウイルスであるとは限らず、DNAウイルスであるB型肝炎ウイルス (HBV)がその一例である。 B型肝炎ウイルスは転写でプレゲノムRNAを生成したのちに逆転写によってDNAを合成している。レトロウイルス科のウイルスは直径約100nmの球状の形をしており、脂質のエンベロープに覆われている。ウイルス粒子のコア(中心)には逆転写酵素と通常2組の同一なRNAゲノムを持つ。共通の遺伝子としては、プロモーター活性のある2つのLTR(long terminal repeat)と呼ばれる塩基配列の繰り返し領域に挟まれて、gag(構造タンパク質をコード)、pro(プロテアーゼをコード)、pol(逆転写酵素などをコード)、env(エンベロープのタンパク質をコード)などの遺伝子を持っている。ウイルスのエンベロープが細胞膜の受容体と結合することで、細胞内にRNAと逆転写酵素が侵入する。その後逆転写酵素が作用し、プラス鎖ウイルスRNAを鋳型にマイナス鎖DNAを合成する。そして合成されたマイナス鎖DNAを鋳型にプラス鎖DNAが合成され、一本鎖RNAが二本鎖DNAに変換される。その後二本鎖DNAは宿主細胞のDNAに組み込まれ、プロウイルスと呼ばれる状態になる。プロウイルスは恒常的に発現している状態となっており、ウイルスRNAやメッセンジャーRNAが次々と合成されていく。メッセンジャーRNAはウイルス蛋白を合成させ、完成したウイルスは宿主細胞から発芽していくのです。
⑦浸潤と転移の活性化という表現(活性化)は間違いです。癌の浸潤とは、ヘルペス完成性がん細胞が原発巣から離脱して周囲の組織や臓器に広がっていく現象です。この浸潤のきっかけとなる癌細胞同士ががっちりスクラムを組んで一体となっている癌細胞塊に一個の癌細胞でヘルペスが最大限増殖するために癌細胞自身が生き続けるために必要な生化学機構やエネルギーや5大栄養素のすべてを利用つくされてしまうと癌細胞も生きられなくネクローシスで死んでしまいます。がん細胞のみならず隣接する癌細胞にはなっていない正常細胞もヘルペスの増殖もためにすべてを利用されすぎたヘルペス感染正常細胞も溶解感染のために細胞が破壊されて壊死してしまいますというよりも餓死してしまいます。因みに末期がんで悪液質で癌患者が癌死といわれる死はヘルペスに金や財産や食い物をすべて無理やり略奪され多細胞の集団的餓死なのです。ある高名な信頼できるある病理学者の書物には末期がんの患者の癌細胞の数は数兆個もあると書かれていました。となればヘルペスウイルスは数十兆個以上も隠れ住んでいることになります。つまり癌死した人や悪液質で死んだ人はすべて膨大な数のヘルペス感染で殺されてしまったのです。
癌転移の始まりとなる癌浸潤はどのようにして開始されるのでしょうか?まず一個の癌細胞が癌細胞も正常細胞も混ざった集団の一体化した細胞組織からはがれ始めるにはまず一個の癌細胞が離脱する必要があるのです。本来一匹のヘルペスウイルスは人間の細胞に感染するのは我が子であるビリオンをできり限り多く作るためにです。ヒトの体内に感染したヘルペスウイルスは一個の正常な細胞に感染しやすい部位は管腔臓器であり特に管腔臓器は上皮組織といわれるのは外部に面した最外層の粘膜上皮細胞なのです。上皮組織の一個の上皮細胞は外側から上皮細胞の層と基底層と結合組織から成り立っています。上皮組織のタイプには3種類あり①単層円柱上皮②重層上皮③腺上皮で腺上皮には外分泌戦細胞と内分泌細胞の2つがあります。だから癌は上皮細胞癌が99%に達するのです。だからヘルペスウイルスが増殖するために癌細胞が増えるために必要な栄養分であるあらゆる種類のアミノ酸や塩類やビタミン類やグルコースや脂質を全部盗まれます。絶対に死ぬことは無いヘルペスが増えれば増えるほど癌細胞は痩せて衰えていくばかりなのに現代の癌専門家は癌細胞が増えれば増えるほど元気溌溂として正常細胞を駆逐していくという間違った考えを持ち続けています。元気溌溂として癌細胞のみならず正常細胞でヘルペスビリオンを細胞が死に絶えるまで増やし切れらの細胞を駆逐していくのはヘルペスウイルスなのです。このように癌細胞のみならず正常細胞で増えたビリオンは感染細胞のゲノムに侵入して癌関連細胞遺伝子である癌原遺伝子と癌抑制遺伝子を好き放題に変異させ益々新し癌細胞を産生するのみならず、すでに癌細胞になった細胞の癌原遺伝子と癌抑制遺伝子の変異の数を増やして癌細胞の悪性化に貢献するのです。癌細胞の悪性化の度合いは800個もあるその癌細胞の癌関連細胞遺伝子が変異して癌化した数によって決まるのです。
病理医が顕微鏡で見た癌細胞の顔つきが悪いので悪性度が強いとよく言いますが癌細胞の顔つきとは何でしょうか?がんの顔つきとは、がん細胞の分化度や異形成の度合いを指す言葉で、病理の専門家が顕微鏡でがんを見たときに用います。がん細胞は正常な役割を果たすために分化しますが、細胞内にある核が大きくなり形がゆがむことで秩序正しく整列できなくなります。この異形成の度合いや違いを「顔つき」と呼び、悪性度の高いがんは「顔つきが悪いがん」と表現されます。がん細胞が、本来の正常な細胞の形態をどれくらい維持しているかを「分化度」といい、「未分化」「低分化」「高分化」などと表現します。分化がんと未分化がんの違いとは?正しい分化の定義はヘルペスウイルスによって
分化とは、分化・未分化とは、もともとは細胞の成長のプロセスをあらわす言葉です。
細胞は分裂をくり返すことで、数を増やしていきますが、さらに分化をすることで、その細胞が属する臓器の働きを持つようになります。未分化がんは、細胞としては未熟な状態にあるといえます。分化がんが、未分化がんへ転化することがあります。それははじめ分化がん
分化がんが進んだ細胞は、急激に育つことはありません。細胞は、分化している度合いが高いほど成熟度が高く、細胞分裂も盛んではないのです。そのため分化がんは、進行が遅く、何年もそのままの状態がつづくことがよくあります。しかし、分化がんは、突然、未分化がんになることがあります。
未分化がんは、正常な甲状腺にいきなりできるわけではなく、長年にわたって存在していた乳頭がんや濾胞がんの性質が変わり、未分化がんに転化するといわれています。
甲状腺がんは、高分化の腺がん(乳頭がん、濾胞がん)から、低分化がんへと段階的に悪性転化し、未分化がんへ転化していくと考えられています。しかしこの文化度という言葉は誤解を生みやすい言葉ですから「細胞の形態の正常性」というべきです。分化度の低いがん細胞は、悪性度が高く活発に増殖する傾向があります。病理検査でがん細胞の分化度を調べることで、悪性度の評価や治療効果の予測などを行います。
異形成とは、細胞が正常では見られない形態になる、形態変化の一種である。通常、上皮組織や造血組織に生じるものをいう。上皮の異形成の病理像については上皮内癌ほどではない構造異型や細胞異型を示す細胞から成る病変で、異型の高度な高度異形成は、前がん病変あるいは良性と悪性の境界病変とされる。通常は体積の増加がみられないものを指す(体積が増加しているものは、「境界悪性腫瘍」などと呼ぶ)。異型度が高度でないものも、しばしば不可逆的に徐々に異型度を増してゆくこともあり、時として悪性腫瘍に進行する。
異形成上皮の細胞周期は正常の上皮より短くなっており、実態としては表層の細胞の脱落のために見かけ上の体積が増えていない上皮性新生物と考えられるが、ただし、表層細胞脱落がアポトーシスのために起こる、すなわちまだ不死化していない細胞であるとしたら、新生物とは言えない。悪性度の高いがんでは早期に転移や再発が起こる可能性があり、薬物治療の選択にも影響します。たとえば、乳がんでは悪性度(グレード分類)によって腋窩リンパ節転移が陰性なのに再発する可能性を予測したり、術後の抗がん剤治療を決めたりします。
がん巣は異なる種類の細胞であるといわれますが組織には正常な多種類の細胞があるので癌になる細胞が異なればがん巣は異なる種類の細胞から成り立っているといえます。しかし癌細胞の種類が異なればん治療が難しい理由は、がん自体が非常に複雑な疾患であるためといわれますが間違いです。何故ならばあらゆるがんを作ったのはヘルペスによる癌関連細胞遺伝子が変異して癌化しただけですから癌を治せる本当の治療はすべて同じであるからです。
細胞のがん化は、唯一の要因はヘルペスだけであり癌関連遺伝子の変異によって1個の癌細胞が生まれます。しかしいつ生まれたかは知る由もありません。出来上がった1個の癌細胞の癌関連細胞遺伝子の変異が積み重ねられると癌の悪性度や顔つきや異形成が高度になるだけです。によって引き起こされます。これにより、細胞増殖の周期で制御されている細胞の成長が制御を失い、異常な速度で増殖し始めるといわれますが実は癌細胞は細胞増殖の周期チェックポイントでは制御できないから癌細胞なのです。細胞周期・チェックポイントとがん
私たちの体を形作るあらゆる細胞は、増殖、分化、あるいは細胞死の機構が適切に働くことで、特定の臓器・組織を正しく構築するための秩序を保っています。一方で、ひとたびこれらの組織構築の秩序が乱されると、がんや奇形、萎縮などの異常が起きます。
細胞周期とは、一つの細胞が分裂して二つの娘細胞に増える過程をいい、上記の組織構築の秩序を保つための重要な制御システムの一つです。細胞周期(図1)には4つのステップ、すなわちDNAの複製を行うS期、細胞分裂を行うM期に加え、それぞれの準備期間であるG1とG2期があります。各ステップで、DNA配列や染色体の異常がある場合に細胞周期をストップさせる「チェックポイント」と呼ばれる機構が存在します。

左図は細胞周期チェックポイントの4つが示されています。
ほとんど全てのがんでは、細胞周期やチェックポイント(監視)の制御システムに異常(特に細胞周期の過剰な活性化やチェックポイントの不具合)が起きており、つまりは細胞周期の異常ががん細胞の特性をサポートしていることが知られています。しかし一方で、それらの異常が、がんの発生や進展に具体的にどう影響しているかは解明されていませんといわれています。が癌は一言でいうとヘルペスによる感染症ですから細胞周期チェックポイントには全く関わりのないことなのです。それに加えて癌細胞が生まれたのは細胞周期チェックポイントの機能がはたらかなくなったからです。上の図で示されているように4つの細胞周期チェックポイントの役割の覚え方はほとんどがDNA損傷があるどうかとDNA複製が問題ないかどうか、つまりせいじょうなDNAを作るかどうかを監視して細胞周期をとめることです。例外が一つありますが分裂してできた新しい二つの細胞の46本の染色体の分配が元の細胞と全く同じかを監視しているのです。ところがこの染色体もDNAの集まりですから4つの細胞周期はわかりやすく「DNAチェックポイント」というべきですね。
細胞周期チェックポイント(DNAチェックポイント)とは、何でしょうか?細胞が正しく新しい二つの同じ細胞を作るために初めから終わりまでの工程である細胞周期を進行させているかどうかを監視(チェック)し、そのこうていの途中で異常や不具合がある場合には細胞周期進行を停止(もしくは減速)させる制御機構のことである。細胞自体がこの制御機構を備えている。一回の2個の新しい細胞を作る細胞分裂の周期の中に、複数のチェックポイントが存在します。下図に示されているようにG1/Sチェックポイント、S期チェックポイント、G2/Mチェックポイント、M期チェックポイントの4つが解析されています。この機構は正確な遺伝情報を娘細胞、ひいては子孫に伝達するための、生命にとって根源的な役割を果たしている。この機構の異常はヒトなどのがん発生の主要な原因のひとつといわれるが実はチェックポイントの異常が癌を作るのではないのです。ヘルペスウイルスが癌原遺伝子を変異させただけですからチェックポイント機構は直接、関わりはありません。

細胞は、その性状や生体内での役割に応じて、それぞれ決まった周期で細胞分裂を繰り返し増殖している。この、一回の分裂増殖の周期を細胞周期と呼び、例えばいくつかの種類のヒト培養細胞の細胞周期は約24時間である。しかし、細胞にX線を照射してDNAに損傷を起こすと、この周期が長くなります。細胞にはDNA損傷などの遺伝子異常が起きると、それを検知して細胞周期を一旦停止させる機構が存在することが発見され、この遺伝子異常を監視し細胞周期を止める機構は細胞周期チェックポイントと名付けられた。略してチェックポイントともいわれる。因みにDNA損傷が癌を作るのではないのです。
細胞周期チェックポイントは、
①遺伝子(DNA)に損傷がないか(DNA損傷チェック)
②DNA複製が正常に行われているか(DNA複製チェック)
③有糸分裂中に、複製された染色体の分離が正しく行われるか(スピンドルチェック)。
などを監視しており、これらに異常が検知されると、チェックポイント制御因子と呼ばれる複数の分子群が活性化されて、細胞周期の進行を遅らせ、停止させる。スピンドルチェックとは紡錘体チェックポイントと訳し、スピンドルチェックポイント、紡錘体形成チェックポイント、有糸分裂チェックポイントとも呼ばれる。有糸分裂または減数分裂において、複製された各染色体が紡錘体に正しく接着するまで染色体の分離を防ぐ、細胞周期チェックポイントである。この間隙を縫ってヘルペスウイルスは染色体の分離の前後に何とか無理やり新しい染色体に侵入して染色体の数の異常や染色体転座などの染色体の不可逆な変異を起こしてしまうのです。
チェックポイント制御因子が活性化されると、その異常の原因が取り除かれるまで、細胞周期が停止した状態になる。この間に、例えば軽度のDNA損傷の場合には、DNA修復機構が働くことで損傷が修復される。そして異常が完全に取り除かれたと検知された時点で、チェックポイントの働きが可逆的に解除され、再び細胞周期が進行する。このように細胞周期チェックポイントは、細胞分裂の過程で異常が生じた場合に、細胞周期を一旦停止させて異常の原因を取り除くことで、遺伝子異常が子孫に伝わらないようにする役割を果たしているのです。
また一方で、重度のDNA損傷の場合などDNA修復機構でも完全な修復が出来ない場合、チェックポイント活性化に続いて、その細胞がアポトーシスを起こして死滅することも明らかになった。この機構は、遺伝子異常を起こした細胞が「自殺」することで、異常な細胞を後世に残さないようする役割を果たしているのです。細胞周期チェックポイントは、その細胞が損傷修復を経て再び増殖に向かうか、アポトーシスを起こすかというスイッチの制御にも関与しています。
チェックポイント機能に異常がおきると、内因性、外因性のDNA損傷によって、細胞は正確な娘細胞(コピー)を作れなくなる場合が多くなる。たとえば、チェックポイント機能の不良により生存に必須な遺伝子に損傷が起きた場合、その細胞は娘細胞を残せずにやがて死滅する。したがって、チェックポイント機能の異常は遺伝情報の正確な伝達において大きな不具合を持つことを意味し、生物にとって重大な脅威となりうる。実際にチェックポイント制御に関与するタンパク質群の変異が起きると、その細胞ではDNA損傷に対する感受性が増して、アポトーシスなどの細胞死を起こしやすくなる。
細胞周期チェックポイントは、そのチェックポイントが細胞周期のどの段階(ステージ)に存在するかによって分類され、これまでに (1) G1/Sチェックポイント、(2) S期チェックポイント、(3) G2/Mチェックポイント、(4) M期チェックポイントの4つが比較的よく解析されている。これらはそれぞれ複数の、異なるチェックポイント制御関連分子によって制御されており、その制御機構は極めて複雑である。
G1/Sチェックポイント
G1期からS期に移行する際のチェックポイント。G1期DNAに損傷がないこと、これからのDNA複製のためのヌクレオチドなどが十分あること、細胞の大きさがチェックされる。多細胞生物では、増殖が許されているか(たとえば、サイトカインがあるか)、増殖が必要な細胞であるか、などもチェックされる。がん抑制遺伝子産物p53、RbとRbのホモログはこの制御を司っているのです。ホモログ(相同遺伝子)とは、進化の過程で共通の祖先から由来する遺伝子を指し、構造や機能において類似性を持つことが多いです。生物学の多くの分野で重要な意味を持ち、遺伝子の進化、機能、および生物間の関係性の解析に利用されます。ホモログと似た言葉に、パラログ:同一ゲノム内の重複した遺伝子とオーソログ:種分岐によって共通の祖先遺伝子から生じた相同な遺伝子の二つがあります。この制御がDNA損傷などで活性化するとS期開始、すなわちDNA複製が阻害され、細胞はG1期にとどまる。酵母などは環境条件が良くない場合や、または人の様な多細胞生物において細胞分裂が適当でない場合、G1停止が長く続くとG0期という休眠状態に入ることもある。G0期ではタンパク質合成が抑制され、細胞周期の進行に関わるタンパク質が一部分解される。
S期チェックポイント
S期のDNA複製の速さを制御し、DNA複製に不具合が検知された場合、複製を遅らせる機構。DNA損傷ではヒトのATM蛋白質はこの制御に関与していると言われる。
G2/Mチェックポイント
G2期からM期に移行する際のチェックポイント。この制御がDNA損傷などで活性化するとM期開始が阻害され、細胞はG2期にとどまる。DNA損傷応答においては、ATR(ataxia-telangiectasia mutated related)がそれ自身かあるいは他の因子によって損傷を認識した後にリン酸化を受け活性化されると、ATRはChk1をリン酸化して活性化する。活性型Chk1はCdc25Aのリン酸化を促進し、Cdc25AによるCdc2の脱リン酸化を阻害するため、Cdc2は高リン酸化された不活性な状態に保たれ、M期に進行せずに細胞周期が停止する。また、活性化されたp53(遺伝子転写因子)は14-3-3s(シャペロン)を転写し、それがリン酸化Cdc25と結合し核外へ排出されるため、Cdc2が不活性なままになる。よってM期進行が抑制される。DNA損傷認識後のATRのリン酸化に関わっている因子は現時点でははっきりしていないが、ヒトがん抑制遺伝子産物BRCA1がその役割を担い、DNA損傷に応答したG2/Mチェックポイントの制御を司っているとも言われている。 DNA複製終了を待たずに、M期が開始する酵母変異株を考慮すると、監視(チェック)期間はS期からG2期にわたる比較的長い期間であると考えられる。
M期チェックポイント
M期(有糸分裂期)の途中にあるチェックポイントで、スピンドルチェック(紡錘体監視)が行われる。M期の細胞では、G2期までのステップ(工程)で複製された対を成す姉妹染色分体が、互いにセントロメア付近でコヒーシン複合体によって架橋結合し、また、このコヒーシンを切断するタンパク分解酵素セパラーゼがセキュリンと結合することで不活性化された状態で存在する。
有糸分裂過程の次のステップとして、細胞の両極から伸びる紡錘糸(微小管)が、それぞれの染色分体のキネトコア(セントロメアの一部)に結合する。一対の染色分体が対称になるよう、正しくかつ同時に、紡錘糸を介して細胞の両極に結合しているかどうかがチェックされる(分裂時の染色体の挙動については、染色体の項も参照)。
分裂後期の染色分体の移動に際しては、ユビキチンリガーゼであるAPC/CがCdc20と結合して活性化することが必要となる。紡錘体が正く形成されると、Cdc20の阻害タンパクであるMad2がCdc20との結合から外れ、APC/Cと結合する。活性化したAPC/Cによって、セキュリン蛋白がユビキチン化され、プロテアソーム依存的に分解されることでセパラーゼが活性化し、染色分体間を架橋するコヒーシン蛋白が切断される。これにより、染色分体は紡錘体極へと移動が可能となる。染色分体が両極から伸びた微小管と等しく結合していないうちは、オーロラキナーゼなどのスピンドルチェックポイントタンパクの監視によってAPC/Cの活性化が阻害され、染色分体の分離を抑制する。
がんと遺伝的不安定性への関連
以下の4つのことから、細胞周期チェックポイント制御の部分的破綻はがんの発生とその後の癌の進行で見られる細胞の無制御な異常増殖の大きなひとつの要因ではないかと推測されているが関係はないのですが一応書いておきます。がんの発生とその後の癌の進行で見られる細胞の無制御な異常増殖はすべて増殖したヘルペスウイルスが細胞の癌関連遺伝子である癌原遺伝子と癌抑制遺伝子の変異を増やせば増やすほど癌は悪性度を増し人が癌で死ぬまで癌細胞を増やし続けるのです。すべての癌の事象はヘルペスが癌関連遺伝子である癌原遺伝子と癌抑制遺伝子の変異を起こし続けるからです。
①主要ながん抑制遺伝子産物p53、Rb、BRCA1は細胞周期チェックポイント制御にも関与する(上記)。これは癌になってしまった後の話しですから意味がないのです。
②多くのがん抑制遺伝子産物はヒトのがんで頻繁に不活性化されており、多くのがんの原因であることが多い。これも癌になった後の話しですから意味はないのです。
③細胞周期チェックポイントは正確な遺伝情報の伝達のための基本的な制御機構であり、その異常は遺伝的不安定性をもたらす。がんは遺伝的不安定性を超えた質の異なる話なのです。
④遺伝的不安定性は多くのがん細胞の主要な特徴である。がん細胞は遺伝的不安定性とは全く関係のないヘルペスウイルスがもたらした癌原遺伝子と癌抑制遺伝子の変異によって起こったヘルペス性感染によってもたらされたのです。

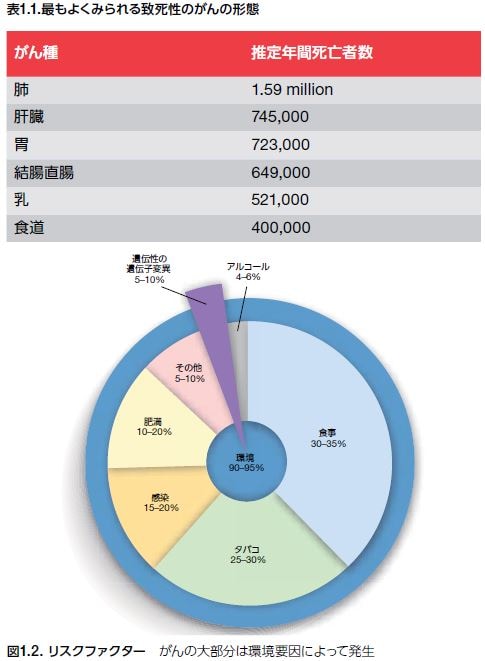{kind=link}
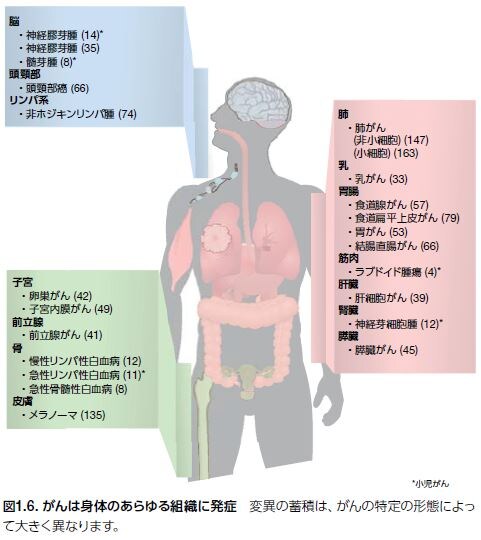{kind=link}
{kind=link}
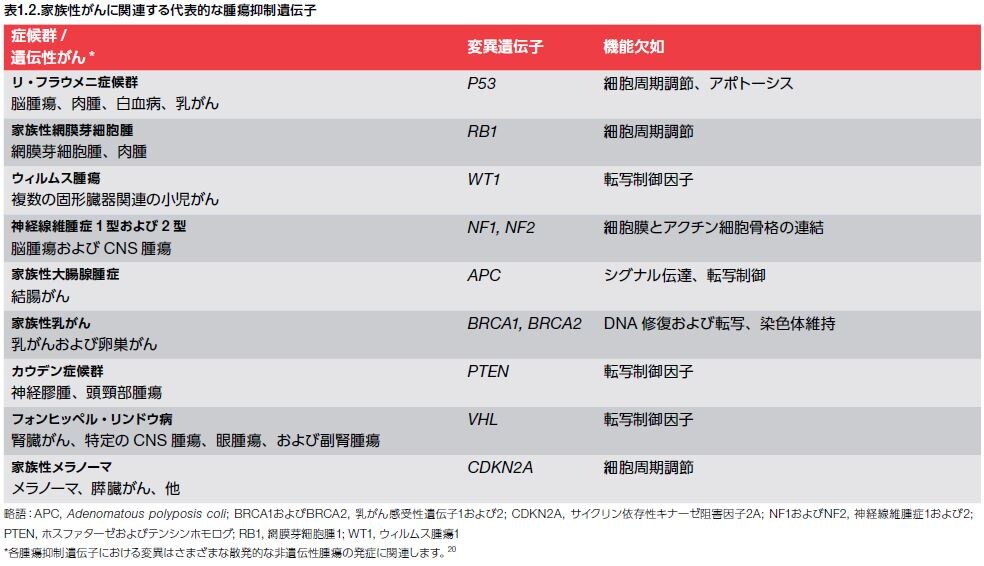{kind=link}
{kind=link}